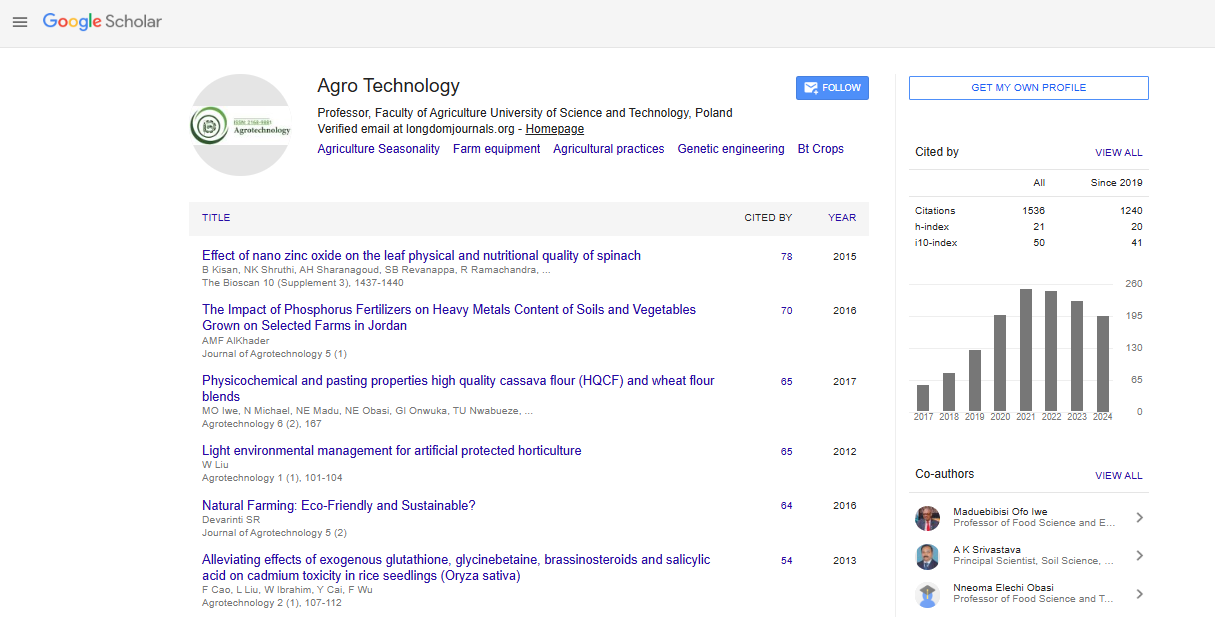

Indexed In
- Open J Gate
- Genamics JournalSeek
- Smithers Rapra
- RefSeek
- Directory of Research Journal Indexing (DRJI)
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Scholarsteer
- Publons
- Geneva Foundation for Medical Education and Research
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2023) Volume 13, Issue 4
The Molecular Docking Study of Interaction of Newly Synthesised Benzamide Appended by Pyrazolone Derivatives as Ligand Molecule with the Target Protein 6LU7 of Novel Corona Virus
Pavithra V1* and Sudha BS22Department of Chemistry, Yuvaraja’s College, Mysore, India
Received: 04-Jul-2023, Manuscript No. ACE-23-21995; Editor assigned: 07-Jul-2023, Pre QC No. ACE-23-21995 (PQ); Reviewed: 24-Jul-2023, QC No. ACE-23-21995; Revised: 31-Jul-2023, Manuscript No. ACE-23-21995 (R); Published: 07-Aug-2023, DOI: 10.35248/2090-4568.23.13.296
Abstract
Plants and bioactive compounds have played an important role in the development of several clinically useful therapeutic agents since time immemorial. The global health emergency of novel COVID-19 is due to severe acute respiratory syndrome corona virus-2 (SARS-CoV-2). As if now there are no approved drugs for the treatment of corona viral disease (COVID-19), although some of the drugs have been tried. The virtual interaction of the COVID-19 main protease in complex with the inhibitor N3 (Research Collaborators for Structural Bioinformatics Protein Data Bank (PDB) ID: 6LU7), Hence this is chosen as target protein molecule. The newly synthesised compounds of benzamide appended pyrazolones derivatives are made as ligand molecules. The synthesis of these organic ligand molecules is done through four steps using different reagents and different environmental conditions. In this article we have discussed the interaction of our synthesized ligands with the target protein molecule using autodock tools. Firstly, the ligands were prepared using commercial ACD/chemsketch tool in PDB format. Desired protein target 6LU7 is downloaded from Protein Data Bank. Further protein optimization done by removing co-ordinates and hetero atoms, energy minimization of protein is done by Swiss PDB viewer 4.1.0. To change the file format open babel 2.4.1 is used. Docking of protein and ligand is done using autodock 4.2 and the results are visualized by pymol and tabulated using the Lamarckian genetic algorithm. After docking comparative study of the binding energies, inhibitory constant and hydrogen bonding of all interactions were discussed and tabulated for the good results.
Keywords
Benzamide; Pyrazolones; Protein-ligand interaction; Molecular docking; Autodock 4.2; Pymol
Introduction
The molecular docking approaches are used to model the interaction between a small molecule and a protein at the atomic level, which allow us to characterize the behavior of small molecules in the binding site of target proteins as well as to elucidate fundamental biochemical processes and study of its features. The docking process involves two basic steps they are prediction of the ligand conformation as well as its position and orientation within these sites (usually referred to as pose) and also assessment of the binding affinity [1].
Pyrazolones are proven to be valuable over wide range of applications in the pharmaceutical industries. The most classic approach to prepare pyrazolones is via the reaction between α, β-ketoester, α, β-cyanoester or α, β-unsaturated esters and hydrazine derivatives of alkanes, arenes and heterocycles. Synthesis of benzamide appended heterocycles was initiated, to expand the application of synthesized compounds as the useful drugs [2].
Plan for synthesis of ligands
Synthesis of substituted benzamides 3a-f was achieved by reacting aminophenol and benzyol chloride in presence of trimethylamine at room temperature for 24 hrs, further synthesis of compounds 4a-f is accomplished by reaction of substituted benzamides 3a-f with ethylbromoacetate in presence of potassium carbonate (Scheme 1). The obtained acetates 4a-f were reacted with 80% hydrazine hydrate to yield substituted hydrazides 5a-f. These phenyl hydrazides 5a-f were used further for cyclisation with ethylacetoacetate in presence of 20% tetrabutylammonium hydroxide in methanol with ethylene glycol as solvent to achieve substituted N-(4-(2-(5-methyl-3-oxo-2,3- dihydropyrazolyl-1-)2-oxoethoxy)phenyl) benzamide derivatives 6a-f which is our ligand molecule (Scheme 2). The structure of synthesized ligands were confirmed by characterization using spectroscopic techniques [3,4].

Scheme 1: Synthesis of ligand molecules
Reagents: i=Dry THF, RT, TEA
ii=Ethylbromoacetate, K2CO3, MeOH
iii=80% Hydrazine hydrate, Methanol
iv=Ethylacetoacetate, TBAH in Methanol, Ethylene glycol, RT

Scheme 2: Structure of benzamide appended by pyrazlones acting as ligands
Chemicals and solvents used were purchased from Sigma Aldrich and used without further purification unless stated. Melting points were determined in open capillaries on a Buchi oil melting point apparatus and are uncorrected Equation(c). Reactions were monitored by using Thin Layer Chromatography (TLC) on aluminum sheet precoated with silica gel 60 F254 (0.2 mm Merck). Chromatographic spots were visualized by UV light and/or with iodine. For column chromatography, silica gel of 100-200 mesh size was used. 1H NMR spectra were acquired on a Brucker NRC-IISC instrument, Banglore at ambient temperature at 400 MHz and/ or 300 MHz in DMSO-d6 or CDCL3 and TMS was used as an internal reference. 13C NMR spectra were recorded on a Bruker AMX-400(100 MHz) with complete proton decoupling. Chemical shift are reported in ppm from TetraMethylSilane (TMS) with the solvent as the internal reference (CDCl3:δ 77.0 ppm or DMSO δ 40.0 ppm). LC-MS was performed on Agilent LCMS system equipped with a BEH C8, 30 × 4.6 mm, 1.7 µm column at a flow rate of 1.0 ml/min [5].
Molecular docking
Molecular docking is a well-established computational technique which predicts the interaction energy between two molecules. This technique is basically incorporates the algorithms like molecular dynamics, fragment search methods and so on. Molecular docking also gives the clear idea that what biochemical mechanism is going on between protein and the ligand. In this article we used to study the interaction of two molecules to fit the best orientation of ligand which would form a complex with overall minimum binding energy [6]. We start with the in-silico generation of ligand and conversion of file format. Further selection of protein and preparation of protein by optimization of protein and energy minimization. Next using Autodock Vina 4.2 autodock was run to check the interaction and finally analysed by visualization tools and creation of algorithm table [7].
Using commercial ACD/chemsketch tool we have drawn the ligand structure and saved in MDL mol.files and further this file is converted into PDB file format by using open babel 2.4.1 which is a necessary format to run autodock. The selected protein is downloaded from RCSB Protein Data Bank in PDB format. In this article we have selected COVID-19 main protease in complex with the inhibitor N3 (Research Collaborators for Structural Bioinformatics Protein Data Bank (PDB) ID: 6LU7. Next the protein optimization and energy minimization is done using swiss PDB viewer 4.1.0. by deleting all hetero atoms and co-ordinates.
Both ligand and protein molecules are prepared in PDB format which is necessary to run autodock. During docking all water molecules are removed from the protein and polar hydrogen atoms and kollman charges were added to the protein and assigned with AD4 types of atoms. Grid selected around 0.512 value was fixed for every run around the active site of protein that is GLN189 which is obtained by literature survey. The docked files are saved in DLG format by Lamarchian Genetic Algorithm and further it is used for visualization and tabulation of results [8-12].
The results of molecular docking and the poses of ligand- protein interaction were studied using autock tools for all interactions such as electrostatic forces, hydrogen bonding, and torsional effects and so on.
Further visualization of the poses of ligand-protein interaction by Pymol. Especially to study the hydrogen bonding interaction of ligand and protein in the selected grid region. The interacted amino acids were tabulated and studied for further. More the hydrogen bonds more will be the stability of the complex thus all six ligands were studied for their hydrogen bonding interaction with the same protein 6LU7. The selection of grid box size is same for all type of ligands used in the molecular docking studies. Totally ten runs were considered for the best pose of ligand-protein interaction among them the lowest binding energy is considered as the best pose. Further the selected interaction pose of ligand and protein were studied under pymol to get the hydrogen bonding interactions (Figures 1A-1F and Figures 2A-2F).

Figure 1: Molecular interaction of ligands with molecules.
Note: 1A: Ligand-a interaction with target protein 6LU7; 1B: Ligand-b interaction with target protein 6LU7;1C: Ligand-c interaction with target protein 6LU7;1D: Ligand-d interaction with target protein 6LU7;1E: Ligand-e interaction with target protein 6LU7; 1F: Ligand-f interaction with target protein 6LU7

Figure 2: Visualization of protein-ligand interaction in terms of hydrogen bonding.
Note: 2A: Visualization of ligand-a interaction with 6LU7; 2B: Visualization of ligand-b interaction with 6LU7; 2C: Visualization of ligand-c interaction with 6LU7; 2D: Visualization of ligand-d interaction with 6LU7; 2E: Visualization of ligand-e interaction with 6LU7; 2F: Visualization of ligand-f interaction with 6LU7
Results and Discussion
Interaction of ligand-a (N-{4-[2-(5-methyl-3-oxo-2, 3-dihydro-1H-pyrazol-1-yl)-2-oxoethoxy] phenyl} benzamide)
After docking the protein target with each of our ligands, we have observed several information regarding the interactions. Each ligand has slight difference in their interaction type with the selected target protein 6LU7 molecule. According to it for ligand-a and protein 6LU7 interaction we found the least binding energy as -8.90 in the 8th run with the inhibition constant of 301.49 nM and the entropy of the selected grid was found to be 0.41. When the complex in viewed in pymol we found that Ligand-a is having three hydrogen bonds with the target protein molecule 6LU7 with the selected grid box. The bond length of the three hydrogen bonds are 2.0, 2.4 and 2.5 with the amino acids GLU166, SER144 and LEU141 respectively [13].
Interaction of ligand-b (4-chloro-N-{4-[2-(5-methyl-3-oxo-2,3-dihydro-1H-pyrazol-1-yl)-2-oxoethoxy]phenyl}benzamide)
Ligand-b and protein 6LU7 interaction we found the least binding energy as -8.77 in the 7th run with the inhibition constant of 374.22 nM and the entropy of the selected grid was found to be 0.11. When the complex in viewed in pymol we found that Ligand-a is having one hydrogen bonds with the target protein molecule 6LU7 with the selected grid box. The bond length of that one hydrogen bonds is 2.0 with the amino acids GLY143 [14].
Interaction of ligand-c (2,4-dichloro-N-{4-[2-(5-methyl- 3-oxo-2,3-dihydro-1H-pyrazol-1-yl)-2-oxoethoxy]phenyl} benzamide)
Ligand-c and protein 6LU7 interaction we found the least binding energy as -8.99 in the 3rd run with the inhibition constant of 255.23 nM and the entropy of the selected grid was found to be 0.14. When the complex in viewed in pymol we found that Ligand-a is having one hydrogen bonds with the target protein molecule 6LU7 with the selected grid box. The bond length of that one hydrogen bonds is 2.9 with the amino acids ARG188 [15].
Interaction of ligand-d (4-bromo-N-{4-[2-(5-methyl-3-oxo-2,3- dihydro-1H-pyrazol-1-yl)-2-oxoethoxy]phenyl}benzamide)
Ligand-d and protein 6LU7 interaction we found the least binding energy as -8.85 in the 7th run with the inhibition constant of 325.66 nM and the entropy of the selected grid was found to be 0.14. When the complex in viewed in pymol we found that Ligand-a is having one hydrogen bonds with the target protein molecule 6LU7 with the selected grid box. The bond length of that one hydrogen bonds is 3.2 with the amino acids GLU166 [16-18].
Interaction of ligand-e (4-methoxy-N-{4-[2-(5-methyl-3- oxo-2,3-dihydro-1H-pyrazol-1-yl)-2-oxoethoxy]phenyl} benzamide)
Ligand-e and protein 6LU7 interaction we found the least binding energy as -8.70 in the 3rd run with the inhibition constant of 420.33 nM and the entropy of the selected grid was found to be 0.10. When the complex in viewed in pymol we found that Ligand-a is having two hydrogen bonds with the target protein molecule 6LU7 with the selected grid box. The bond length of that two hydrogen bonds is 2.3 and 4.8 with the amino acids GLY143 and GLN189 respectively [19,20].
Interaction of ligand-f (N-{4-[2-(5-methyl-3-oxo-2,3-dihydro- 1H-pyrazol-1-yl)-2-oxoethoxy]phenyl}-4-nitrobenzamide)
Ligand-f and protein 6LU7 interaction we found the least binding energy as -9.17 in the 8th run with the inhibition constant of 190.41 nM and the entropy of the selected grid was found to be 0.15. When the complex in viewed in pymol we found that Ligand-a is having three hydrogen bonds with the target protein molecule 6LU7 with the selected grid box. The bond length of that three hydrogen bonds is 2.1, 2.3 and 2.1 with the amino acids GLN192, GLN192 and GLY143 respectively (Figure 3A-3F) (Table 1).

Figure 3: Pymol viewer of hydrogen bondings in the protein-ligand complex.
Note: 3A: Ligand-a interaction with 6LU7 protein through hydrogen bonding; 3B: Ligand-b interaction with 6LU7 protein through hydrogen bonding; 3C: Ligand-c interaction with 6LU7 protein through hydrogen
bonding; 3D: Ligand-d interaction with 6LU7 protein through hydrogen bonding; 3E: Ligand-e interaction with 6LU7 protein through hydrogen
bonding; 3F: Ligand-f interaction with 6LU7 protein through hydrogen bonding
| Name of ligand | Best | Binding energy | Inhibition constant ki | Entropy of cluster | Number of hydrogen bonds | Bond length | Interacted amino acid |
|---|---|---|---|---|---|---|---|
| Run | |||||||
| Ligandâ??a | 8 | -8.9 | Â 301.49 nM | 0.41 | 3 | 2 | GLU166 |
| 2.4 | SER144 | ||||||
| 2.5 | LEU141 | ||||||
| Ligandâ??b | 7 | -8.77 | 374.22 nM | 0.11 | 1 | 2 | GLY143 |
| Ligandâ??c | 3 | -8.99 | 255.23 nM | 0.14 | 1 | 2.9 | ARG188 |
| Ligandâ??d | 7 | -8.85 | 325.66 nM | 0.14 | 1 | 3.2 | GLU166 |
| Ligandâ??e | 3 | -8.7 | 420.33 nM | 0.1 | 2 | 2.3 | GLY143 |
| 4.8 | GLN189 | ||||||
| Ligandâ??f | 8 | -9.17 | 190.41 nM | 0.15 | 3 | 2.1 | GLN192 |
| 2.3 | GLN192 | ||||||
| 2.1 | GLY143 |
Table 1: Comparative study of interaction of ligand molecule with target protein 6LU7.
Conclusion
More the number of hydrogen bond in the complex more will be the stability and lesser the value of inhibition constant (Ki) more will be the interaction power of the ligand with protein which means a small concentration of ligand can show a good interaction with the protein molecule. When compare the interaction results of all the ligands with same selected protein target molecule, ligand-f is having a good interaction with protein when compare to all ligands because ligand-f is having binding energy as -9.17 in the 8th run with the inhibition constant of 190.41 nM.
Declaration
Ethical approval
The data and visualization images used in this research article are not plagiarized by anywhere.
Competing interests
Not applicable
Author’s contribution
The whole article is written and data are obtained by author Smt. Pavithra V and the guidance for the research was provided by the corresponding author Dr. Sudha B S.
Funding
For this research article no funding is utilised and no funding agencies are involved.
Availability of data and materials
The data base and software used for this research article are reproducible and available for universal usage.
ORCID ID
Pavithra V
Orcid id: https://orcid.org/0000-0003-4209-9830
References
- Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et.al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020; 579(7798):270-273.
[Crossref] [Google Scholar] [PubMed]
- Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. Lancet. 2020; 395(10223):470-473.
[Crossref] [Google Scholar] [PubMed]
- Elmancy L, Alkhatib H, Daou A. SARS-CoV-2: an analysis of the vaccine candidates tested in combatting and eliminating the COVID-19 virus. Vaccines. 2022; 10(12):2086.
[Crossref] [Google Scholar] [PubMed]
- McConkey BJ, Sobolev V, Edelman M. The performance of current methods in ligand–protein docking. Current Science. 2002; 10:845-56.
- Jorgensen WL. The many roles of computation in drug discovery. Science. 2004; 303(5665):1813-1818.
[Crossref] [Google Scholar] [PubMed]
- Bajorath J. Integration of virtual and high-throughput screening. Nat Rev Drug Discov. 2002; 1(11):882-894.
[Crossref] [Google Scholar] [PubMed]
- Langer T, Hoffmann RD. Virtual screening an effective tool for lead structure discovery. Curr Pharm Des. 2001; 7(7):509-527.
[Crossref] [Google Scholar] [PubMed]
- Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat Rev Drug Discov. 2004; 3(11):935-949.
[Crossref] [Google Scholar] [PubMed]
- Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov. 2020; 19(3):149-50.
[Crossref] [Google Scholar] [PubMed]
- Lim J, Jeon S, Shin HY, et al. Case of the Index Patient Who Caused Tertiary Transmission of COVID-19 Infection in Korea: The Application of Lopinavir/Ritonavir for the Treatment of COVID-19 Infected Pneumonia Monitored by Quantitative RTPCR. J Korean Med Sci. 2020; 35:e79.
[Crossref] [Google Scholar] [PubMed]
- Lesser R, Weiss R. Über selenhaltige aromatische Verbindungen (VI). Berichte der deutschen chemischen Gesellschaft (A and B Series). 1924; 57(7):1077-1082.
- Müller A, Cadenas E, Graf P, Sies H. A novel biologically active seleno-organic compound—1: Glutathione peroxidase-like activity in vitro and antioxidant capacity of PZ 51 (Ebselen). Biochem Pharmacol. 1984; 33(20):3235-3239. [Crossref]
[Google Scholar] [PubMed]
- Nakamura Y, Feng Q, Kumagai T, Torikai K, Ohigashi H, Osawa T, et.al. Ebselen, a glutathione peroxidase mimetic seleno-organic compound, as a multifunctional antioxidant: Implication for inflammation-associated carcinogenesis. J Biol Chem. 2002; 277(4):2687-2694.
[Crossref] [Google Scholar] [PubMed]
- Saito I, Asano T, Sano K, Takakura K, Abe H, Yoshimoto T, et.al . Neuroprotective effect of an antioxidant, Ebselen, in patients with delayed neurological deficits after aneurysmal subarachnoid hemorrhage. Neurosurgery. 1998; 42(2):269-277.
[Crossref] [Google Scholar] [PubMed]
- Yamaguchi T, Sano K, Takakura K, Saito I, Shinohara Y, Asano T, et.al. Ebselen in acute ischemic stroke: A placebo-controlled, double-blind clinical trial. Stroke. 1998; 29(1):12-17.
[Crossref] [Google Scholar] [PubMed]
- Imai H, Graham DI, Masayasu H, Macrae IM. Antioxidant Ebselen reduces oxidative damage in focal cerebral ischemia. Free Radic Biol Med. 2003; 34(1):56-63.
[Crossref] [Google Scholar] [PubMed]
- Parnham M, Sies H. Ebselen: Prospective therapy for cerebral ischaemia. Expert opinion on investigational drugs. 2000; 9(3):607-619.
[Crossref] [Google Scholar] [PubMed]
- Wendel A, Fausel M, Safayhi H, Tiegs G, Otter R. A novel biologically active seleno-organic compound—II: Activity of PZ 51 in relation to Glutathione Peroxidase. Biochem Pharmacol. 1984; 33(20):3241-3245.
[Crossref] [Google Scholar] [PubMed]
- Haenen GR, De Rooij BM, Vermeulen NP, Bast AA. Mechanism of the reaction of Ebselen with endogenous thiols: Dihydrolipoate is a better cofactor than glutathione in the peroxidase activity of ebselen. Mol Pharmacol. 1990; 37(3):412-422.
[Google Scholar] [PubMed]
- Masumoto H, Kissner R, Koppenol WH, Sies H. Kinetic study of the reaction of ebselen with peroxynitrite. FEBS Lett. 1996; 398(2-3):179-182.
[Crossref] [Google Scholar] [PubMed]
Citation: Pavithra V, Sudha BS (2023). The Molecular Docking Study of Interaction of Newly Synthesised Benzamide Appended by Pyrazolone Derivatives as Ligand Molecule with the Target Protein 6LU7 of Novel Corona Virus. Adv Chem Eng. 13:296.
Copyright: © 2023 Pavithra V, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.