
Indexed In
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- JournalTOCs
- ResearchBible
- Electronic Journals Library
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Proquest Summons
- SWB online catalog
- Virtual Library of Biology (vifabio)
- Publons
- MIAR
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
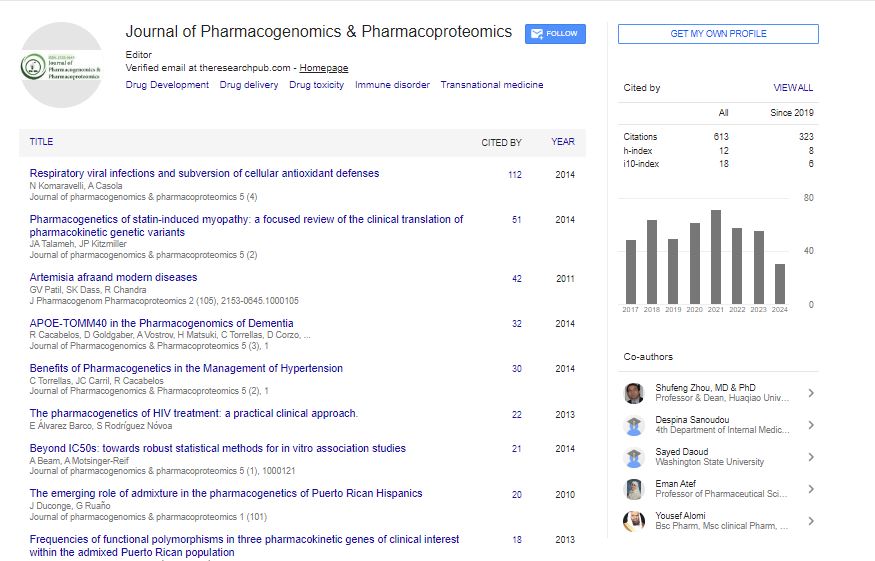
Research Article - (2022) Volume 13, Issue 4
The First Glimpse of Homo sapiens Hereditary Fusion Genes
Degen Zhuo*Received: 06-Jun-2022, Manuscript No. JPP-22-17030; Editor assigned: 09-Jun-2022, Pre QC No. JPP-22-17030 (PQ); Reviewed: 23-Jun-2022, QC No. JPP-22-17030; Revised: 30-Jun-2022, Manuscript No. JPP-22-17030 (R); Published: 07-Jul-2022, DOI: 10.35248/2153-0645.22.13.016
Abstract
Family-inherited fusion genes have been known to be associated with human disease for decades. However, only a small number of them have been discovered so far. This report uses monozygotic (MZ) twins as a genetic model to investigate human hereditary fusion genes (HFGs). We have analyzed RNA- Seq from 37 MZ twins and discovered 1,180 HFGs, the maximum of which is 608 per genome. Based on these data, a human genome encodes at least 1,000 HFGs. We have found that forty-eight HFGs, whose recurrent frequencies are ≥ 25%, are associated with MZ twin inheritance, eight of which are detected in ≥ 52% of 74 MZ twins. Tandem gene duplications and SCO2 gene amplification generate four and two of these eight HFGs, respectively, and, in turn, provide the best and direct scientific self-support for the concept of hereditary fusion genes. Interestingly, two of these eight HFGs are previously-studied cancer fusion genes, which support that they are inherited from parents and not from somatic genomic alteration. Hence, HFGs are major genetic factors for human diseases and complex traits. More importantly, HFGs provide one of the best and most straightforward tools to study genomic alterations in human genetics. This study gives us the first glimpse of human HFGs and lays technological and theoretical foundations for future genetic, biological, and medical studies.
Keywords
Hereditary; Epigenetic; Fusion gene; Monozygotic twins; Inheritance; RNA-Seq; Tandem duplication; Gene amplification; Genomic alteration
Introduction
A gene was thought to be a unit of inheritance that ferried a characteristic from parent to child [1]. Fusion genes such as BCR-ABL1 have been traditionally thought to be somatic and cancerous [2,3] and, hence, not hereditable [4]. Human family-inherited fusion genes generated by genomic alterations were responsible for significant inherited pathology of humans (Homo sapiens) [5-7]. Human germline genomic structural variants (SV) are the genetic foundation of hereditable fusion genes [8]. Limitations available to genome technologies historically hindered accurate SV identification [9,10]. As genome technologies progressed from array-comparative genomic hybridization to long-read sequencing and other emerging technologies, the prevalence of human genomic SVs has dramatically increased from about 300 to 34,234 SVs per human haploid genome [10]. However, these complex SVs were often mapped to multiple locations in a genome, making it impossible to obtain reproducible data for genetic studies [7]. During the last several decades, traditional molecular cloning has been extensively used for investigating SV-associated human diseases, such as human color blindness, which was discovered in 1998 [11]; inherited peripheral neuropathies [12]; and other diseases [12-16].
Recent advances in RNA-Seq technologies have identified large numbers of fusion transcripts, most of which have been thought to be somatic and cancerous [17]. On the other hand, many studies have shown that many fusion transcripts exist in high frequencies in non-cancer tissues [18-20]. In addition to read-through fusion transcripts [20], fusion transcripts resulting from genomic alterations were first discovered in cancer but later in healthy samples. TPM4-KLF2 [21], PIM3-SCO2 [22,23], NCO2-UBC [24], and OAZ1-KLF2 [21] are first reported in cancer samples, the first three of which are later observed in normal controls [24]. These apparent contradictions suggested that fusion genes require further exploitation. Recently, RNA-Seq has been used to identify fusion transcripts associated with rare inherited diseases [25]. Previously, we developed SCIF (splicingcodes identify fusion gene transcripts) to more accurately and efficiently discover fusion transcripts from RNA-Seq datasets and identified enormous numbers of fusion transcripts [26]. However, when we systematically validated cancer-specific fusion genes such as KANSARL (KANSL1-ARL17A), they were detected in healthy samples and individuals at very high frequencies [26]. Eventually, the KANSARL gene was validated as the predisposition (hereditary) fusion genes [26]. To study family- inherited fusion genes more precisely, we defined the hereditary fusion gene (HFG) as the fusion gene that offspring inherited from parents and excluded read-through fusion transcripts generated via transcriptional termination failure. Since environmental and physiological factors regulated read-through [20,27,28], we defined the epigenetic fusion gene (EFG) as the fusion genes generated via cis-splicing of read-through pre-mRNAs of two same- strand neighbor genes of the human reference genome. The main differences between HFGs and EFGs were that HFGs were much younger and generated by human germline genomic alterations after the divergence between human and chimpanzees. This report used monozygotic (MZ) twins, who share identical genetic materials [29], as a genetic model to study human HFGs systematically.
Materials and Methods
Materials
RNA-Seq dataset of monozygotic (MZ) twins: Raw RNA-Seq data of monozygotic (MZ) twins (dbGap-accession: phs000886) was downloaded from NCBI (https://www.ncbi.nlm.nih.gov/Traces/ study/?acc=SRP061248). This dataset contained RNA-Seq data from 37 pairs of monozygotic (MZ) twins’ blood samples.
RNA-Seq dataset of Genotype-Tissue Expression (GTEx): To evaluate the MZ twins’ hereditary fusion genes, we selected the RNA-Seq dataset of GTEx healthy blood samples as a control. RNA-Seq datasets (dbGap-accession: phs000424.v7.p2) of GTEx’s blood samples were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000424.v7.p2). We had identified 427 healthy individual blood samples.
Methods
Identification of fusion transcripts by SCIF (Splicing Codes Identify Fusion Transcripts): SCIF (Splicingcodes Identify Fusion Transcripts) was described previously by Zhou et al. [26].
Classifications of types of fusion transcripts
To better characterize fusion transcripts, fusion transcripts were classified into the following five types based on locations and distances of 5’ and 3’ genes of fusion transcripts.
• Inter-Chromosomal: If 5’ and 3’ genes of a fusion transcript were located on two different chromosomes, the fusion transcript was inter- chromosomal.
• Deletion: If 5’ and 3’ genes of a fusion transcript originated from the identical chromosomes, the distances between the 5’ and 3’ genes were larger than ≥ 250,000 bp, and the 5’ and 3’ genes had identical orientations, the fusion transcript was classified as a deletion.
• Inversion: If 5’ and 3’ genes of a fusion transcript were mapped to opposite strands of the identical chromosomes or if opposite directions of the same chromosomal stands and the distances between the 5’ and 3’ genes were ≤ 250,000bp, the fusion transcript was defined as an inversion.
• Intra-Chromosomal: If 5’ and 3’ genes of a fusion transcript originated from the identical chromosomes and distances between the 5’ and 3’ genes were longer than 10,00,000 bp, the fusion transcript was defined as intra-chromosomal.
• Read-Through: If 5’ and 3’ genes of a fusion transcript were on the identical chromosomal strands and had the same directions and the distances between the 5’ and 3’ genes were smaller than 250,000 bp, the fusion transcript was classified as a read-through.
Identification of hereditary fusion genes (HFGs): We specifically defined hereditary fusion genes (HFGs) as the fusion genes offspring inherited from parents and excluded epigenetic (read- through) fusion genes. Fusion genes were defined as chimeric genes originating from two different genes whose distances were ≥ 1,000,000bp. This study used monozygotic (MZ) twins to develop a genetic model to distinguish the somatic fusion genes and hereditary fusion genes. Since MZ twins shared identical genetic materials [29], if a random SV mutation to generate a fusion gene per individual had a rate of 3.6 × 10-2 [30,31], the probability that a pair of MZ twins had the identical SV mutations would be 1.3 × 10-3 and was twenty-seven fold less. Therefore, we could use the probability difference to remove a somatic fusion gene. If a fusion gene had been detected in both MZ twins’ individuals (bHFG), this gene had a frequency of 2.7% (1 out of 37), which was 20-fold higher than the random chance of 1.3 × 10-3. This difference is statistically significantly higher.
If a bHFG had been found in ≥ 1 pair of MZ twins and this bHFG was found in one individual of another pair of MZ twins (iHFG), the chance of this iHFG that was generated due to a random SV mutation was 4.7 × 10-5. Therefore, one iHFG was 1/72 or 0.0139, statistically significantly higher than 4.7 × 10-5. Therefore, this iHFG was counted as a hereditary fusion gene (HFG).
Identification of epigenetic fusion genes (EFGs): Epigenetic fusion genes (EFGs) had been defined as the fusion genes generated via cis-splicing of read- through pre-mRNAs of two same-strand neighbor genes. If the distance of the two same-strand neighbor genes were ≤ 250,000 bp long, the new fusion gene from these two genes was the EFG. Since gene orders and the genomic structures were highly conserved in a species or even among different species, the read- through pre-mRNA was due to failed transcriptional terminations and regulated by environmental and physiological factors [20,27,28]. Therefore, we defined the genes to produce the read-through products as EFGs. Healthy individuals had almost identical EFG genomic sequences, and EFGs were frequently detected. EFG expression patterns may be different among different tissues and developmental stages.
Recurrent frequency of hereditary fusion genes (HFGs): To calculate a recurrent frequency of an HFG, the observed number of samples having the HFG was divided by the total number of samples.
Results
Discovery of human hereditary fusion genes (HFGs) using monozygotic twins’ RNA-Seq
Since MZ twins share identical genetic materials and even identical epigenetics [29], Figure 1a showed that an identical HFG (indicated by ‘H’) was carried by a fertilized egg and inherited by two identical embryos. If a random SV mutation (indicated by ‘S’ in Figure 1a) generated a somatic fusion gene, it would be detectable only in one of the MZ twin siblings [32]. If a random SV mutation to generate a fusion gene per individual had a rate of 3.6 × 10-2 [30,31], the probability that two identical MZ twins had a random somatic fusion gene was 1.3 × 10-3. The probability that both individuals of an MZ twin had an identical HFG was 1/37 or 2.7% and 20-fold higher than that of the random somatic mutations. We used SCIF to analyze blood RNA-Seq data from 37 pairs of MZ twins (dbGap accession: phs000886) and identified 97,770 fusion transcripts. From these fusion transcripts, we identified a total of 1180 HFGs, shown in Supplementary Table 1, in both siblings of ≥ 1 pair of MZ twins (bHFG), whose frequencies range from 1 to 23 pairs of MZ twins (Figure 1b). This MZ 1180 bHFGs counted for only 1.2% of the total fusion transcripts and 15.2% of 7,750 fusion transcripts detected in ≥ 2 individuals (Supplementary Table 1), suggesting that the MZ bHFGs were not due to random chances. Two hundred seventy-one (23%) of 1180 HFGs had been observed in ≥ 2 pairs of MZ twins (Figure 1b), the average of which was 3.97 pairs of MZ twins. Hundreds of HFGs present in multiple pairs of MZ twins mathematically ruled out that these HFGs were generated via random somatic genomic alterations. In addition, Figure 1c showed that 946 (80.2%) of 1,180 bHFGs had been found to have 1–18 HFGs present in one of two MZ twin siblings (iHFG), the average of which was 4.88 iHFGs. If a bHFG existed in ≥ 1 pair of MZ twins, the chance of its iHFG generated via a random ‘S’ mutation was ≤ 4.7 × 10-5, which was at least 46 fold less than the observed iHFG frequencies, which ranged from 1.4% to 25%. iHFG was equal to its counterpart of the bHFG and would be treated as the HFG unless specified. To get each individual’s total HFGs, we added bHFGs and iHFGs together (Supplementary Table 2). On average, each of 1180 HFGs was detected in 7.3 persons or 9.86% of the 74 MZ twin individuals, which was statically significantly higher than the ‘S’ random mutation [30,31]. Therefore, the fusion gene ‘H’ was the hereditary fusion gene offspring inherited from their parents.

Figure 1: Brief review and characterization of identification of hereditary fusion genes (HFGs). a). schematic diagrams to show the formation of two monozygotic (MZ) twin siblings from a monozygote. ‘H’ and ‘S’ represented the hereditary fusion gene and a random somatic genomic alteration. b). recurrent gene frequencies of bHFGs among 37 pairs of MZ twins. bHFGs were fusion genes detected in both siblings of an MZ twin. c). recurrent gene frequencies of iHFGs among 1180 HFGs. iHFGs were fusion genes detected in one individual of an MZ twin among 1180 HFGs. e) Venn diagram showing overlapped HFGs between KN6790 and KN4750 MZ twins. Light blue and white circles represented KN4750 and KN6790 MZ twins, respectively. f) Venn diagram displayed overlapped MZ HFGs and the fusion transcripts discovered in 427 GTEx blood samples. Dark green and white circles showed MZ HFGs and the fusion transcripts discovered in 427 GTEx blood samples. g). Venn diagram showed the overlapped numbers of fusion transcripts between 37 pairs of MZ twins and 427 GTEx blood samples. Light blue and white circles showed MZ fusion transcripts and the fusion transcripts discovered in 427 GTEx blood samples.
Next, we analyzed the HFG distribution among the MZ twins’ siblings. Figure 1d showed that MZ twins ranged from 21 HFGs in KN650 MZ twins to 608 HFGs in KNAA4 MZ twins. The average MZ twin individual encoded 178.8 HFGs (Supplementary Table 2). Since we show that numbers of fusion transcripts correlate with high-quality RNA-Seq data and RNA-Seq data sizes [26], the enormous differences among different MZ twin individuals might be mainly due to RNA-Seq qualities. Supplementary Table 3 showed that KN650 and KNAA4 MZ twins had similar RNA-Seq data sizes. KNAA4 MZ twins, SRR2105729 and SRR2105730, had 6212 and 7153 fusion transcripts from which 498 and 510 HFGs, respectively, were identified, while KN650 MZ twins, SRR2105720 and SRR2105721, had 82 and 163 fusion transcripts from which three and 18 HFGs, respectively, were found (Supplementary Table 4). Therefore, the actual number of HFGs would be significantly higher, suggesting that HFGs were widespread and highly diverse among different individuals.
To demonstrate HFG complexities, we selected and compared KN4750 and KN6709 MZ twins. Supplementary Table 2 showed that KN4750 and KN6709 had 426 HFGs and 348 HFGs. A comparison of KN4750 and KN6709 showed that the 180 HFGs overlapped and accounted for 51.7% of KN4750’s 348 HFGs and 42.3% of KN6790’s 426 HFGs (Figure 1e). To generate the overlapping 180 HFGs, KN4750 and KN6709 were expected to have 673 and 1007 HFGs, respectively. Potential 1007 HFGs encoded by KN6709 confirmed that human genomes encoded large numbers of HFGs. Hence, they, in turn, created genotypic and phenotypic diversities.
To confirm that human HFGs were conserved and widespread, we investigated whether these HFGs existed in the Genotype-Tissue Expression (GTEx) fusion genes. We used SCIF to analyze 427 GTEx blood samples (dbGap-accession: phs000424.v7.p2) and identified 134,090 fusion transcripts. Figure 1f showed that 576 (48.8%) of 1180 HFGs were present in the total fusion genes found in GTEx’s blood samples. On the other hand, Figure 1g showed that 7,989 fusion transcripts were found in both MZ twins and GTEx’s blood samples and accounted for only 8.7% of the total MZ twins’ fusion transcripts. The former was more than fivefold higher than the latter, confirming that the probabilities of these HFGs inherited by the MZ twins were conserved and had significantly higher frequencies in general populations than other fusion transcripts. Supplementary Table 5 showed that these 576 HFGs were present in 420 GTEx blood samples and ranged from 0.2% to 40.1%, while the MZ twins’ counterparts ranged from 1.4% to 67.7%, the average of which was 10.4%. The former average frequency was 1.4% and was sevenfold less than the MZ twins’ counterpart, reflecting genetic differences between the two populations. Supplementary Table 6 showed that 98.4% of 427 GTEx samples had 1 to 37 HFGs, and the average was 7.9 HFGs, supporting the fact that HFGs were conserved, extremely diverse, and widespread.
Characterization of potential mechanisms of generating human hereditary fusion genes (HFGs)
To understand the potential mechanisms of generating these diverse HFGs, we arbitrarily classified HFGs into five groups: within-a-gene inversion, inversion, deletion, intra-chromosomal fusion genes, and inter-chromosomal fusion genes. Figure 2a showed that LIMS1-LIMS1 was a within-a-gene inversion, more likely to be generated via a direct LIMS1 gene tandem duplication. Since SCIF had deliberately removed highly repetitive sequences, identifying within-a-gene inversion HFGs was due to gene homologs and pseudogenes. Hence, the numbers of within-a-gene inversion may be significantly underestimated. Figure 2b showed that head-to-tail MEG8- SNOR114 genes were rearranged into SNOR114-MEG8 gene structure by inversion to produce a novel non-coding RNA HFG. Figure 2c showed that PLEKHO1 and ANP32E were located on 1q21 opposite strands to form a tail-to-tail structure, and a potential inversion of the PLEKHO1 gene might generate head-to-tail ANP32E-PLEKHO1 HFG. Figure 2d showed that a potential deletion of sequences between TPM4 and KLF2 might form TPM4-KLF2 HFG detected in 54.1% of 74 MZ twins. Figure 2e showed that potential intra-chromosomal translocation produced a RORA-B2M HFG. Figure 2f showed that a potential inter-chromosomal translocation resulted in the generation of the OAZ1-SCO2 HFG. Inter-chromosomal alterations generated 660 HFGs, which counted for 55.9% of 1180 HFGs and met theoretical expectations. As shown in Figure 2, the potential mechanisms to generate HFGs were not different from those observed in somatic genomic alterations, suggesting that the generation of HFGs was a classical genetic event in the germline cells [8]. Therefore, unless it was under natural selection, any potential fusion gene generated by germline structural variants was a potential HFG and had a much higher frequency than its somatic counterpart if its inheritability was not impaired.

Figure 2: Schematic diagrams of potential genomic alterations for producing human hereditary fusion genes (HGFs). a). Within-a-gene inversion HFG, which was generated via tandem gene duplications. b). MEG8 was inverted to SNORD114-1 on chromosome 14q32. c). Upstream PLEKHO1 was inverted to downstream of ANP32E on chromosome 1p21 to generate ANP32E-PLEKHO1. d) The DNA sequences were deleted between TPM4 and KFL2 genes to produce TPM4-KFL2. e) B2M was translocated downstream of RORA on chromosome 15q21 to generate RORA-B2M. f). SCO2 on chromosome 22q13 was translocated downstream of OAZ1 on chromosome 19p13 to form the OAZ1-SCO2 fusion gene. Red, black, and gray horizontal arrows represented the 5’ gene, 3’ gene, and genes surrounding both genes. Horizontal arch arrows showed genomic alterations to produce fusion genes.
To increase SCIF computation speed, we intentionally removed repetitive DNA sequences and most intergenic sequences. To understand the potential roles of repetitive DNAs, we used the TRNAN35 gene, coding for transfer RNA asparagine 35, as an example to illuminate HFG generation and potential roles during evolution. Figure 3a showed that the TRNAN35 gene, located at 1q21, was inverted to the positive strand upstream of the SRGAP2P gene to form a TRNAN35-SRGAP2P HFG. Figures 3b and 3c showed that TRNAN35 was translocated to the regions upstream of FAM91A3P and ZNF238 genes to produce TRNAN35-FAM91A3P and TRNAN35-ZNF238 HFGs, respectively. Figures 3d, 3e, 3f and 3g showed that TRNAN35 was translocated into different chromosomes to yield four putative TRNAN35-fused HFGs. Interestingly, TRNAN35-SRGAP2P, TRNAN35-FAM91A3P, TRNAN35-ACTB, TRNAN35-CHD2, and TRNAN35-UBBwere detected in the individual SRR2105730 blood sample. TRNAN35 could co-regulate the expression of these five HFGs via interactions with aspartyl/glutamyl-tRNA(Asn/Gln) amidotransferase. Hence, they may form a natural network regulated by TRNAN35. The addition or deletion of TRNAN35-fused HFGs would dramatically increase network diversity and biological diversities. ALU-SINE exonization was the extreme example, which increased protein diversity [33,34] and provided regulatory networks [35] to human genetic and biological diversity.

Figure 3: Using the TRNAN35 gene as a model to demonstrate that repetitive DNA sequences played essential roles in forming hereditary fusion genes (HFGs). a).TRNAN35 was inverted to upstream of SRGAP2P on chromosome 1q21.2 to generate TRNAN35-SRGAP2P; b).TRNAN35 was translocated upstream of FM91A3P on chromosome 1q21.1 to form TRNAN35-FAM91A3P; c).TRNAN35 was translocated upstream of ZNF238on chromosome 1q44 to yield TRNAN35-ZNF238; d).TRNAN35 was translocated upstream of ACTB on chromosome 7q15 to form TRNAN35- ACTB; e).TRNAN35 was translocated upstream of CHD2 on chromosome 15q26 to produce TRNAN35-CHD2; f).TRNAN35 was translocated upstream of SEPT9 on chromosome 17q25.2 to form TRNAN35-SEPT9; g).TRNAN35 was translocated upstream of UBB on chromosome 17p12 to generate TRNAN35-UBB. Green arrows were TRNAN35 and 5’ genes. Solid red and gray arrows represented 3’ genes and the genes surrounding TRNAN35 and 5’ genes, respectively. Horizontal combos of the white arch and solid green arrows showed TRNAN35 translocations to produce fusion genes.
Identification and characterization of hereditary fusion genes (HFGs) associated with monozygotic twins’ inheritance
Family genetic analysis shows that MZ twin inheritance is family-inherited, but no genetic factors have been discovered so far [36]. Hence, we explored whether HFGs were associated with MZ twins’ genetics. We identified 50 HFGs, ranging from 25% to 67.6%, and 48 were associated with MZ twin inheritance. We could not determine whether KANSARL (KANSL1-ARL17A) detected in 27% of MZ twin siblings was not associated with MZ twin inheritance due to unknown racial mixes. Table 1 showed that eight HFGs were detected in over 52.7% of 74 MZ twins’ individuals, ranging from 52.7% to 67.6%, while the GTEx counterparts ranged from zero to 5.2%. The formers were statistically significantly higher than the latter, suggesting that the MZ twins’ inheritance was a complex trait. Half of the eight HFGs, including LIMS1-LIMS1, SDHAP2- SDHAP2, POM121C-POM121C, and PLEKHM1-PLEKHM1 were within-a-gene inversions and originated from tandem gene duplications (Table 1). SDHAP2-SDHAP2 was a pseudogene tandem duplication, while LIMS1-LIMS1, POM121C-POM121C,and PLEKHM1-PLEKHM1 were protein-coding gene tandem duplications.
| Fusion Gene ID | FT Types | GTEx Blood (427) | MZ Twins (74) | ||
|---|---|---|---|---|---|
| # of HFGs | % | # of individuals | % | ||
| PLXNB2-SCO2 | INVERSION | 10 | 2.4 | 50 | 67.6 |
| LIMS1-LIMS1 | INVERSION | 2 | 0.5 | 50 | 67.6 |
| SDHAP2-SDHAP2 | INVERSION | 0 | 0 | 44 | 59.5 |
| BACH1-MECP2 | INTER_CHR | 21 | 5 | 43 | 58.1 |
| TPM4-KLF2 | DELETION | 4 | 1 | 40 | 54.1 |
| POM121C-POM121C | INVERSION | 2 | 0.5 | 40 | 54.1 |
| PLEKHM1-PLEKHM1 | INVERSION | 2 | 0.5 | 40 | 54.1 |
| PIM3-SCO2 | INVERSION | 22 | 5.3 | 39 | 52.7 |
Note: # of HFGs for GTEx Blood; #of individuals for MZ Twins
Table 1: Eight hereditary fusion genes (HFGs) detected in ≥ 50% of 74 MZ twin siblings. The GTEx blood samples were used as a healthy control. The numbers inside the brackets indicated sample sizes.
LIMS1-LIMS1 was one of the most frequently detected HFGs associated with MZ twin inheritance, and LIMS1 tandem duplication (Figure 2a) added two extra exons. It encoded a truncated LIM and senescent cell antigen-like domains 1 (Supplementary Figure 1), which is likely involved in integrin signaling through its LIM domain-mediated interaction with integrin-linked kinase. POM121C- POM121C HFG added two extra exons to 5’ UTR of the POM121C gene encoding POM121 membrane glycoprotein C (Supplementary Figure 2). Sequence analysis showed that adding two exons resulted in no protein sequence change. Similarly, PLEKHM1- PLEKHM1 fusion added two extra exons to 5’ UTR of PLEKHM1 coding for pleckstrin homology domain-containing family M member 1 (Supplementary Figure 3a). Sequence analysis showed that adding two extra exons produced a new open read frame of 250 a.a protein (Supplementary Figure 3b), with 97% of sequence identity with human pleckstrin homology domain-containing family M member 1 isoform X4 (Supplementary Figure 3c). The rest were TPM4-KLF2, BACH1-MECP2, PLXNB2-SCO2, and PIM3-SCO2. TPM4-KLF2 HFG was a deletion fusion gene between the TPM4 gene, encoding tropomyosin 4 and KLF2 gene, coding for kruppel-like transcription factor 2 and producing a fusion gene encoding a putative 97 aa fusion protein (Supplementary Figure 4).
Table 1 showed that PLXNB2-SCO2 and PIM3-SCO2 were two SCO2-fused HFGs. PLXNB2, PIM3, and SCO2 genes were from the 22q13 genomic region (Figure 4a).

Figure 4: Potential genomic alterations on chromosome 22q13.33 to generate SCO2-fused HFGs. a). The schematic diagram showed the 22q13.33 genomic structure. b). SCO2 was translocated downstream of PLXNB2 to generate PLXNB2-SCO2; c) SCO2 was translocated downstream of PPP6R2 to produce PPP6R2-SCO2; d) SCO2 was translocated downstream of TRABD to form TRABD-SCO2; e) SCO2 was translocated to downstream of PIM3 to generate PIM3-SCO2. Solid red arrows were SCO2 genes. Solid black and gray arrows represented 5’ genes and the genes surrounding the SCO2 and 5’ genes. White arrows indicated genomic alterations to generate the SCO2-fused fusion genes.
PLXNB2-SCO2 was one of the most recurrent HFGs and was detected in 67.6% of 74 MZ twin siblings. PLXNB2-SCO2 was an inversion fusion gene between PLXNB2, encoding plexin B2 and SCO2, coding for cytochrome C oxidase assembly protein (Figure 4b). PLXNB2 gene brought a new translation initiation codon for the PLXNB2-SCO2 HFG and increased SCO2 protein by ten amino acids (Supplementary Figure 5). PIM3-SCO2 was also an inversion fusion gene between the PIM3 gene, encoding the PIM-3 oncogene and SCO2 gene. PIM3-SCO2 resulted in a frame shift and shortened PIM3 protein by 120 amino acids (Supplementary Figure 6). Further inspections showed that PPP6R2-SCO2 and TRABD-SCO2 were also from the 22q13 genomic region (Figure 4c-4e) and detected in 41.9% and 28.4%, respectively. PPP6R2-SCO2 was an inversion fusion gene between PPP6R2 encoding protein phosphatase 6 regulatory subunit 2 and SCO2 gene and encoded a putative 737 a.a. PPP6R2-SCO2 hybrid protein (Supplementary Figure 7). TRABD-SCO2 was a fusion gene between TRABD, encoding TraB domain-containing protein, and the SCO2 gene. TRABD gene furnished the TRABD-SCO2 HFG with a promoter and 5’ UTRs and resulted in no change of SCO2 protein. Supplementary Table 6 showed that the recurrent frequencies of these GTEx’s SCO2- fused HFGs ranged from 2.1% to 5.2% and were significantly less frequent than the MZ ones, suggesting these four SCO2-fused HFGs were associated with the inheritance of MZ twins. Nearly half of MZ twin siblings had two SCO2-fused HFGs. Furthermore, some twin individuals, such as SRR2105686 and SRR2105716, had all four SCO2-fused HFGs, suggesting that SCO2-fused HFGs resulted from amplifying the SCO2 gene. Supplementary Table 1 showed that an additional eleven SCO2-fused HFGs were present in 1180 HFGs, the highest OAZ1-SCO2 detected in 24.3% of the MZ twins (Figure 2f). These SCO2-fused HFG data supported the SCO2 gene amplification. Since they were classic examples of studying human genetics, tandem gene duplications and amplification provided the best and most direct scientific self-support for the notion of hereditary fusion genes.
Discussion
This report used MZ twins as a genetic model to identify 1180 HFGs from 37 pairs of MZ twins. The maximum numbers of HFGs were 608 per genome. To generate the overlapped HFGs among different groups of MZ twins, we found that MZ genomes encoded over 1000 HFGs. Because a human genome was shown to have 34,234 SVs [10], a genome might encode thousands of HFGs. As shown in Figure 3, one gene could fuse multiple genes to generate multiple HFGs. Therefore, each gene could generate new fusion genes with every other human gene. If the human genome encoded 25,000 genes [37], each of which had nine exons [38], human genomes could generate 5×109 HFGs during evolution. As Figure 2 shows, genomic alterations resulting HFGs were genomic amplification and tandem duplications (Figure 4). They maintained typical gene structures and had fewer impacts on their heritability except for gene dosage changes. We can predict that offspring could inherit every fusion gene produced via germline genomic alterations unless its inheritability was impaired. Since most single nucleotide variants (SNVs) were located in non-coding regions, they might play only minor roles in human genetics. Hence, hereditary fusion genes were the dominant genetic factors supported by up to 34,234 SVs per human haploid genome [10]. Since hereditary and epigenetic fusion genes significantly increased numbers of fusion transcripts, it suggested that gene numbers, locations, and orders were significant for human genetics.
We had identified 50 HFGs whose recurrent frequencies ranged from 25% to 67.6%, among which eight HFGs were larger than 50%. Half of eight HFGs whose recurrent frequencies were ≥ 50% were fusion genes generated by tandem gene duplication. These four HFGs from tandem gene duplication produced fusion genes and had diverse potential biological functions. SDHAP2-SDHAP2 was pseudogene tandem duplication, while POM121C-POM121Cresulted in tandem duplication of two 5’ UTR exons and no change of the POM121C protein sequences (Supplementary Figure 2). LIMS1-LIMS1 tandem duplication resulted in a frameshift, produced early termination codons, and encoded a truncated LIMS1 protein (Supplementary Figure 1). On the other hand, PLEKHM1- PLEKHM1 was a tandem duplication of two 5’ UTR exons of the PLEKHM1 gene. Sequence analysis showed that PLEKHM1- PLEKHM1 resulted in a new open reading frame (Supplementary Figure 3), which shared 97% of sequence identity with human pleckstrin homology domain-containing family M member 1 isoform X4 (Supplementary Figure 3). Similarly, local amplification of the SCO2 gene at the 22q13 genomic region resulted in four HFGs: PLXNB2-SCO2, PIM3-SCO2, PPP6R2-SCO2, and TRABD-SCO2. TRABD-SCO2 and PLXNB2-SCO2 HFGs added zero and ten a.a. to the N-terminal of SCO2 protein. PIM3-SCO2 HFG resulted in a truncated PIM3 protein (Supplementary Figure 6), while PPP6R2-SCO2 produced a putative PPP6R2-SCO2 hybrid protein (Supplementary Figure 7). Tandem gene duplications (Figure 2a) and amplifications (Figure 2 and Figure 4) were the most common genetic variants. They provided the most direct scientific evidence that hereditary fusion genes were more widespread than expected. Since potential functions of these HFGs were deduced based on the fusion junctions of their main isoforms, the entire length cDNAs of these HFGs had to be characterized to get more accurate information in the future.
Among the eight HFGs detected ≥ 50% of MZ twin siblings, TPM4- KLF2 [21] and PIM3-SCO2 [22,23] are cancer fusion genes. TPM4-KLF2 is first reported in acute myeloid leukemia (AML) [21]. PIM3- SCO2 is discovered in chronic neutrophilic leukemia and children AML [22,23]. Locher et al. show that TPM4-KLF2 is detected in 30% of acute myeloid leukemia samples [24]. However, they have also reported it was present in all three normal bone marrow samples [24], which was divergent from 0.95% of 427 GTEx blood samples, suggesting it’s no random distributions. Previously reported NCO2- UBC [24] and OAZ1-KLF2 [21,24] were HFGs associated with MZ twin inheritance and were detected in 44.6% and 33.8% of 74 MZ twin siblings, respectively. Since TPM4-KLF2, PIM3-SCO2, NCO2- UBC, and OAZ1-KLF2 were detected in 15, 15, 12, and 5 pairs of 37 MZ twins, respectively, the chances of these HFGs generated by random genomic alterations were the maximum of 3.6 × 10-15, suggesting that it was mathematically impossible for these HFGs to be generated by random somatic genomic rearrangements.
Conclusion
Hence, offspring must inherit their parents’ TPM4-KLF2, PIM3- SCO2, NCO2-UBC, and OAZ1-KLF2. These hinted that many originally-thought fusion genes were HFGs. Driver oncogenes and other random fusion genes were generated via somatic genomic alterations in later stages of cancer development. Fusion genes produced by random genomic alterations would have much lower recurrent frequencies than those HFGs. If they were authentic, they would lead paradigm shifts in all aspects of cancer studies including the cancer biology. These data suggested that HFGs were the dominant genetic factors associated with many phenotypes and complex traits from MZ twin inheritance to cancer. Recent advances in genome technologies made it possible to map genomic SVs and validate HFGs directly at the same time to exploit human hereditary fusion genes further. It would help us develop more efficient technologies to uncover more HFGs and discover associations between HFGs and diseases and complex traits.
Acknowledgments
We have expressed our most profound appreciation to Ms. Xiaoyan Yang, Prof. Benoit Chabot, Prof. Jeff Xiwu Zhou, Prof. Shunbin Ning, Prof. Yinxiong Li, Dr. Liren Tang, Mr. David Zhuo, and Mr. Noah Zhuo for their various contributions to successfully transform the SplicingCodes theory to the technologies to validate the hereditary and epigenetic fusion genes during the last two decades.
Disclosures
No authors declare relavant conflict of interest.
REFERENCES
- Pearson H. Genetics: What is a gene? Nature. 2006; 441(7092):398-401.
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007; 7(4):233-245.
[Crossref] [Google Scholar] [PubMed]
- Jia Y, Xie Z, Li H. Intergenically Spliced Chimeric RNAs in Cancer. Trends Cancer.2016; 2(9):475-484.
[Crossref] [Google Scholar] [PubMed]
- Wu H, Li X, Li H. Gene fusions and chimeric RNAs, and their implications in cancer. Genes Dis. 2019; 6(4):385-390.
[Crossref] [Google Scholar] [PubMed]
- Mazzarella R, Schlessinger D. Pathological consequences of sequence duplications in the human genome. Genome Res. 1998; 8(10):1007-1021.
[Crossref] [Google Scholar] [PubMed]
- Puig M, Casillas S, Villatoro S, Caceres M. Human inversions and their functional consequences. Brief Funct Genomics. 2015; 14(5):369-379.
[Crossref] [Google Scholar] [PubMed]
- Eichler EE. Genetic Variation, Comparative Genomics, and the Diagnosis of Disease. N Engl J Med. 2019; 381(1):64-74.
[Crossref] [Google Scholar] [PubMed]
- Pocza T, Grolmusz VK, Papp J, Butz H, Patócs A, Bozsik A. Germline Structural Variations in Cancer Predisposition Genes. Front Genet.2021; 12:634217.
[Crossref] [Google Scholar] [PubMed]
- Huddleston J, Eichler EE. An Incomplete Understanding of Human Genetic Variation. Genetics.2016; 202(4):1251-1254.
[Crossref] [Google Scholar] [PubMed]
- Ho SS, Urban AE, Mills RE. Structural variation in the sequencing era. Nat Rev Genet. 2019; 21(3):171-189.
[Crossref] [Google Scholar] [PubMed]
- Borg MD, Deeb SS, Motulsky AG. Molecular patterns of X chromosome-linked color vision genes among 134 men of European ancestry. Proc Natl Acad Sci.1989; 86(3):983-987.
[Crossref] [Google Scholar] [PubMed]
- Reiter LT, et al. A recombination hotspot responsible for two inherited peripheral neuropathies is located near a mariner transposon-like element. Nat Genet. 1996; 12(3):288-297.
[Crossref] [Google Scholar] [PubMed]
- Hayashi T, Motulsky AG, Deeb SS. Position of a 'green-red' hybrid gene in the visual pigment array determines colour-vision phenotype. Nat Genet. 1999; 22(1):90-93 .
[Crossref] [Google Scholar] [PubMed]
- Bi W, Park SS, Shaw CJ, Withers MA, Patel PI, Lupski JR. Reciprocal crossovers and a positional preference for strand exchange in recombination events resulting in deletion or duplication of chromosome 17p11.2. Am J Hum Genet. 2003; 73(6):1302-1315.
[Crossref] [Google Scholar] [PubMed]
- Higgs DR, Vickers MA, Wilkie AO, Pretorius IM, Jarman AP, Weatherall DJ. A review of the molecular genetics of the human alpha-globin gene cluster. Blood. 1989; 73(5):1081-1104.
[Crossref] [Google Scholar] [PubMed]
- Quinlan AR, Hall IM. Characterizing complex structural variation in germline and somatic genomes. Trends Genet. 2012; 28(1):43-53.
[Crossref] [Google Scholar] [PubMed]
- Mitelman F, Johansson B, Mertens F, Schyman T, Mandahl N. Cancer chromosome breakpoints cluster in gene-rich genomic regions. Genes Chromosomes Cancer. 2019; 58(3):149-154.
[Crossref] [Google Scholar] [PubMed]
- Chase A, et al. TFG, a target of chromosome translocations in lymphoma and soft tissue tumors, fuses to GPR128 in healthy individuals. Haematologica. 2010; 95(1): 20- 26.
[Crossref] [Google Scholar] [PubMed]
- Babiceanu M, et al. Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res. 2016; 44(6):2859-2872.
[Crossref] [Google Scholar] [PubMed]
- Thierry Mieg D, Thierry Mieg J. AceView: A comprehensive cDNA- supported gene and transcripts annotation. Genome Biol. 2006; 7(Suppl 1): S12 1-14.
[Crossref] [Google Scholar] [PubMed]
- Roberts KG, et al. Genetic alterations activating kinase and cytokine receptor signaling in highrisk acutelymphoblastic leukemia. Cancer Cell. 2012; 22(2): 153-166.
[Crossref] [GoogleScholar] [PubMed]
- Kuhn J, et al. PIM3-SCO2 Fusion Is a Novel Transcription-Induced Chimera That Is Highly Prevalent In Childhood AML. Blood. 2013; 122(21):2549-2549.
- Menezes J, et al. CSF3R T618I co-occurs with mutations of splicing and epigenetic genes and with a new PIM3 truncated fusion gene in chronic neutrophilic leukemia. Blood Cancer J. 2013; 3(11): e158-e158.
[Crossref] [Google Scholar] [PubMed]
- Irene J, Locher WA, Daniel M, Borràs M, Willy Honders, Rick H, et al. Fusion Transcripts without Corresponding Cytogenetic Abnormalities in Acute Myeloid Leukemia: Implications for AML Pathogenesis. Blood. 2017; 130(S1): 2703.
- Oliver GR, et al. A tailored approach to fusion transcript identification increases diagnosis of rare inherited disease. PLoS One. 2019; 14(10):e0223337.
[Crossref] [Google Scholar] [PubMed]
- Zhou JX, et al. Identification of KANSARL as the first cancer predisposition fusion gene specific to the population of European ancestry origin. Oncotarget. 2017; 8(31): 50594-50607.
[Crossref] [Google Scholar] [PubMed]
- Ehrlich J, Sankoff D, Nadeau JH. Synteny conservation and chromosome rearrangements during mammalian evolution. Genetics. 1997; 147(1):289-296.
[Crossref] [Google Scholar] [PubMed]
- Ahituv N, Prabhakar S, Poulin F, Rubin EM, Couronne O. Mapping cis- regulatory domains in the human genome using multi-species conservation of synteny. Hum Mol Genet. 2005; 14(20):3057-3063.
[Crossref] [Google Scholar] [PubMed]
- Van Baak TE, et al. Epigenetic supersimilarity of monozygotic twin pairs. Genome Biol. 2018; 19(1): 2.
[Crossref] [Google Scholar] [PubMed]
- Conrad DF, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010; 464(7289):704-712.
- Campbell CD, Eichler EE. Properties and rates of germline mutations in humans. Trends Genet. 2013; 29(10):575-584.
[Crossref] [Google Scholar] [PubMed]
- Nishioka M, et al. Identification of somatic mutations in monozygotic twins discordant for psychiatric disorders. NPJ Schizophr. 2018; 4(1):7.
[Crossref] [Google Scholar] [PubMed]
- Krull M, Brosius J, Schmitz J. Alu-SINE exonization: En route to protein- coding function. Mol Biol Evol. 2005; 22(8):1702-1711.
[Crossref] [Google Scholar] [PubMed]
- Schwartz S, Mark NG, Kfir N, Oren R, Kim E, Ast G. Alu exonization events reveal features required for precise recognition of exons by the splicing machinery. PLoS Comput Biol. 2009;5(3):e1000300 .
[Crossref] [Google Scholar] [PubMed]
- Shen S, et al. Widespread establishment and regulatory impact of Alu exons in human genes. Proc Natl Acad Sci USA. 2011; 108(7):2837-2842.
[Crossref] [Google Scholar] [PubMed]
- Machin G. Familial monozygotic twinning: A report of seven pedigrees. Am J Med Genet C Semin Med Genet. 2009; 151 C(2):152-154.
[Crossref] [Google Scholar] [PubMed]
- Pertea M, et al. CHESS: A new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 2018; 19(1):208.
[Crossref] [Google Scholar] [PubMed]
- Sakharkar MK, Chow VT, Kangueane P. Distributions of exons and introns in the human genome. In Silico Biol. 2004; 4(4):387-393.
[Google Scholar] [PubMed]
Citation: Zhuo D (2022) The First Glimpse of Homo sapiens Hereditary Fusion Genes. J Pharmacogenom Pharmacoproteomics. 13:016.
Copyright: © 2022 Zhuo D This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.