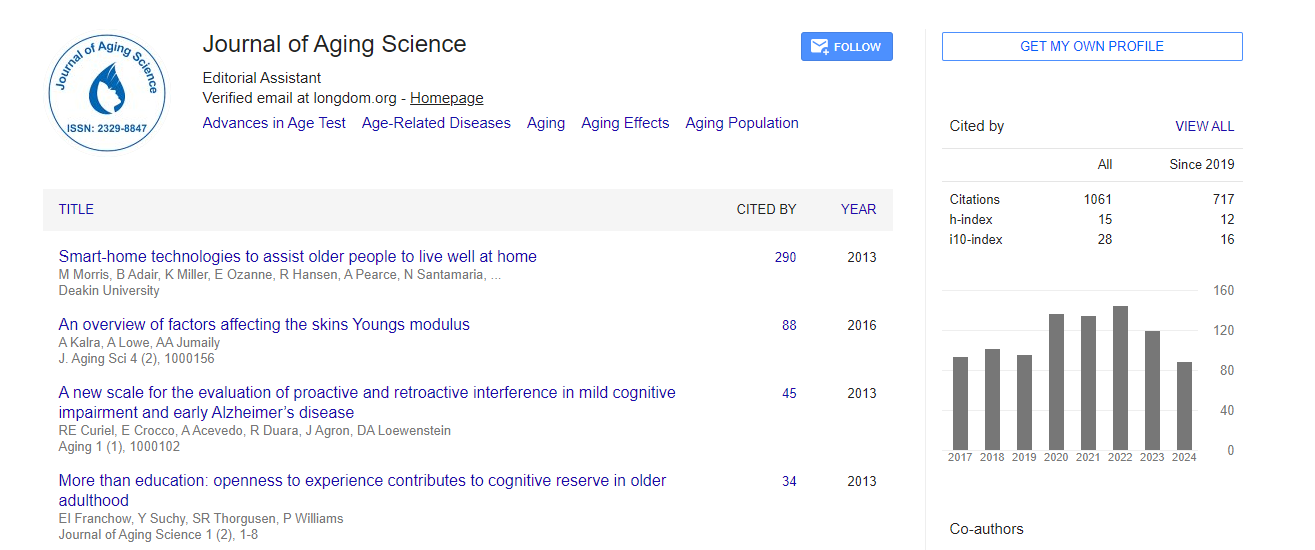
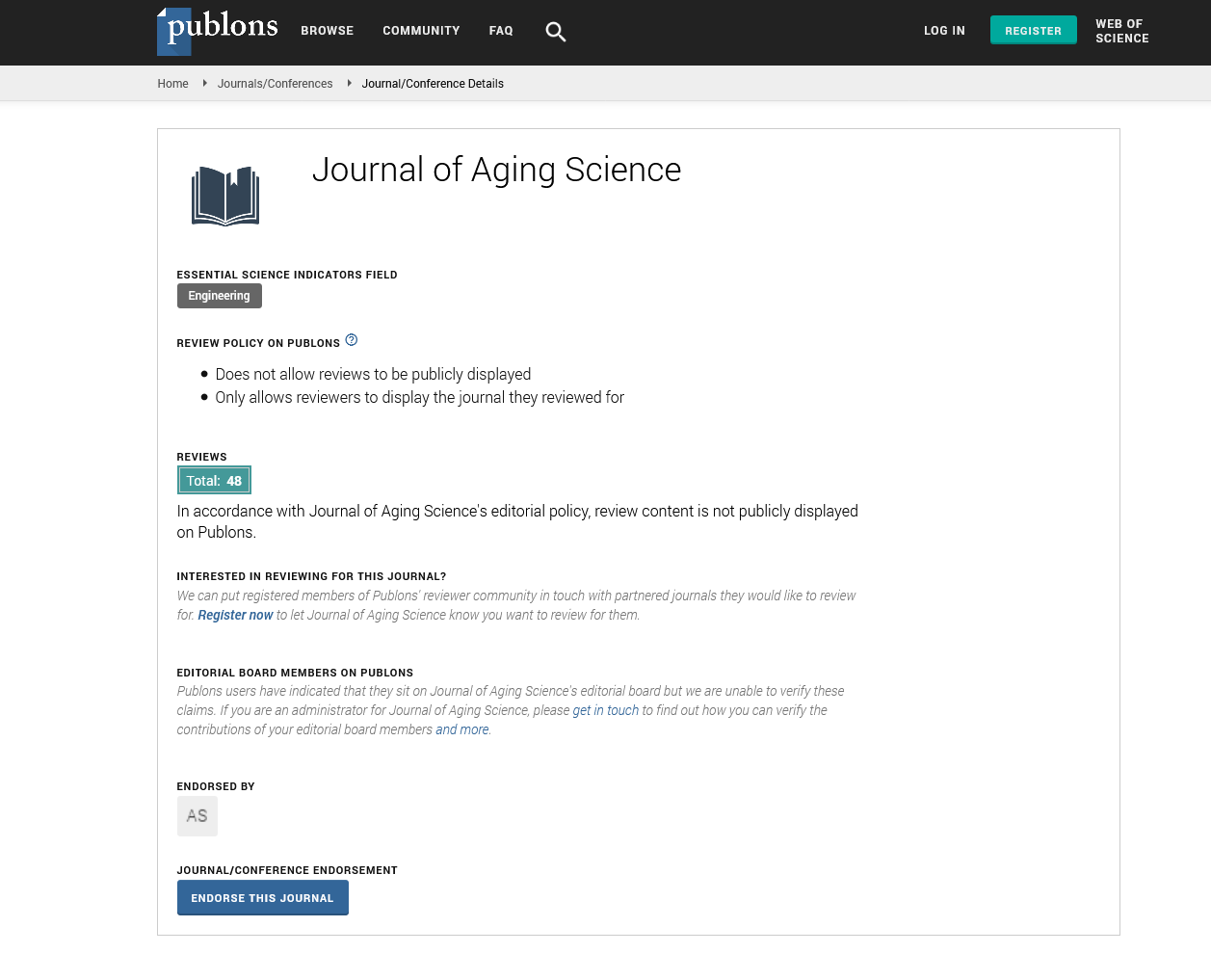

Indexed In
- Open J Gate
- Academic Keys
- JournalTOCs
- ResearchBible
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Opinion - (2025) Volume 13, Issue 2
Microglia Dysfunction and Its Contribution to Neurodegeneration
Adam Beyer*Received: 28-Mar-2025, Manuscript No. JASC-25-28628; Editor assigned: 31-Mar-2025, Pre QC No. JASC-25-28628 (PQ); Reviewed: 07-Apr-2025, QC No. JASC-25-28628; Revised: 14-Apr-2025, Manuscript No. JASC-25-28628 (R); Published: 28-Apr-2025, DOI: 10.35248/2329-8847.25.13.412
Description
Neurodegenerative diseases, including Alzheimer’s Disease (AD), Parkinson’s Disease (PD), Huntington’s Disease (HD) and Amyotrophic Lateral Sclerosis (ALS), are characterized by progressive neuronal loss and cognitive or motor dysfunction. While the primary causes of these disorders vary, accumulating evidence suggests that neuroinflammation plays an important role in disease progression. Central to this inflammatory response are microglia, the resident immune cells of the Central Nervous System (CNS).
Microglia play a dual role in brain health they support normal neuronal function by clearing debris and regulating synaptic plasticity but can also contribute to neurodegeneration when dysregulated. Dysfunctional microglia fail to maintain homeostasis, leading to chronic neuroinflammation, synaptic damage and neuronal death. Understanding microglial dysfunction and its contribution to neurodegeneration is necessary for developing targeted therapies for these debilitating diseases.
Microglia and their functions in brain health
Microglia are specialized macrophage-like cells that reside in the CNS and perform critical immune and homeostatic functions. These cells continuously monitor the brain microenvironment, responding to injury, infection and cellular debris. In a healthy brain, microglia maintain homeostasis through several key functions:
Surveillance and phagocytosis: Microglia constantly extend and retract their processes to survey the CNS for signs of damage or infection. When needed, they remove apoptotic cells, damaged synapses and protein aggregates via phagocytosis.
Synaptic pruning: During brain development and throughout life, microglia help refine neural circuits by selectively eliminating weak or unnecessary synapses, a process important for cognitive function and memory formation.
Neurotrophic support: Microglia secrete neurotrophic factors such as Brain-Derived Neurotrophic Factor (BDNF), which supports neuronal survival and synaptic plasticity.
Regulation of neuroinflammation: In response to injury or infection, microglia release cytokines and chemokines that recruit other immune cells to modulate inflammation.
Under normal conditions, microglia maintain a balance between their protective and inflammatory roles. However, when this balance is disrupted, they can become overactive or dysfunctional, contributing to neurodegenerative diseases.
Microglial dysfunction in neurodegeneration
Microglial dysfunction is an attribute of many neurodegenerative diseases. This dysfunction manifests in two primary forms: excessive activation, leading to chronic inflammation and impaired phagocytic function, resulting in the accumulation of toxic protein aggregates.
Chronic microglial activation and neuroinflammation: In response to pathological stimuli, microglia shift to a proinflammatory state, releasing excessive levels of cytokines such as Tumor Necrosis Factor-alpha (TNF-α), Interleukin-1 beta (IL-1β) and Interleukin-6 (IL-6). While acute inflammation can be beneficial for clearing infections and debris, chronic inflammation leads to sustained neuronal damage.
Alzheimer’s Disease (AD): In AD, microglia become overactivated in response to Amyloid-beta (Aβ) plaques. Instead of efficiently clearing these plaques, they release proinflammatory cytokines, increasing neuronal injury and tau pathology. Studies show that prolonged microglial activation promotes synaptic loss and cognitive decline.
Parkinson’s Disease (PD): In PD, microglia overreact to misfolded alpha-synuclein aggregates, leading to excessive production of Reactive Oxygen Species (ROS) and inflammatory mediators. This results in the progressive loss of dopaminergic neurons in the substantia nigra, a key feature of the disease.
Amyotrophic Lateral Sclerosis (ALS): In ALS, microglia adopt a neurotoxic phenotype, releasing inflammatory molecules that accelerate motor neuron degeneration.
Alzheimer’s Disease (AD): In AD, dysfunctional microglia fail to efficiently clear amyloid-beta plaques, allowing them to accumulate and form neurotoxic deposits. Genetic mutations in microglial-related genes, such as TREM2 (triggering receptor expressed on myeloid cells 2), have been linked to impaired phagocytosis and an increased risk of AD.
Parkinson’s Disease (PD): Microglia in PD show reduced clearance of alpha-synuclein aggregates, which contributes to Lewy body formation and neuronal death.
Huntington’s Disease (HD): Microglial dysfunction in HD leads to inefficient clearance of mutant huntingtin protein, worsening neuronal toxicity and disease progression.
Molecular mechanisms of microglial dysfunction
Several molecular pathways contribute to microglial dysfunction in neurodegenerative diseases.
TREM2 signaling: TREM2 is a receptor expressed on microglia that regulates phagocytosis and inflammation. Mutations in TREM2 impair microglial clearance of Aβ, increasing AD risk.
NF-κB pathway: The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway regulates microglial inflammatory responses. Chronic activation of this pathway leads to sustained neuroinflammation and neurodegeneration.
NLRP3 inflammasome: The NLRP3 inflammasome is a protein complex that activates inflammatory responses in microglia. Overactivation of NLRP3 contributes to AD, PD and ALS pathology.
Oxidative stress and mitochondrial dysfunction: Microglia exposed to chronic stressors produce excessive ROS, leading to oxidative damage and further impairing their function.
Therapeutic strategies targeting microglial dysfunction
Given the role of microglia in neurodegeneration, targeting their dysfunction presents a potential methods for therapeutic intervention.
Non-Steroidal Anti-Inflammatory Drugs (NSAIDs): Some studies suggest that NSAIDs can reduce neuroinflammation and lower the risk of AD.
Minocycline: This antibiotic has shown potential in reducing microglial activation and neurotoxicity in PD and ALS.
Enhancing microglial phagocytosis
TREM2 activation: Strategies to enhance TREM2 signaling may improve microglial clearance of toxic aggregates in AD.
Colony-Stimulating Factor 1 Receptor (CSF1R) inhibitors: These drugs can modulate microglial activation and promote a neuroprotective phenotype.
Modulating the gut-brain axis: Emerging evidence suggests that gut microbiota influence microglial function. Probiotics and dietary interventions targeting the gut-brain axis may offer novel strategies for preventing microglial dysfunction in neurodegenerative diseases.
Conclusion
Microglial dysfunction is a critical contributor to neurodegeneration, playing a central role in diseases such as AD, PD, HD and ALS. While microglia are necessary for maintaining brain health, chronic activation and impaired phagocytosis can drive neuroinflammation and neuronal loss. Understanding the molecular mechanisms behind microglial dysfunction has opened new methods for therapeutic interventions, including anti-inflammatory drugs, phagocytosis enhancers and gut microbiome modulation. As research continues, targeting microglial dysfunction may become a key strategy in developing treatments for neurodegenerative diseases, ultimately improving patient outcomes and quality of life.
Citation: Beyer A (2025). Microglia Dysfunction and Its Contribution to Neurodegeneration. J Aging Sci. 13:412.
Copyright: © 2025 Beyer A. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.