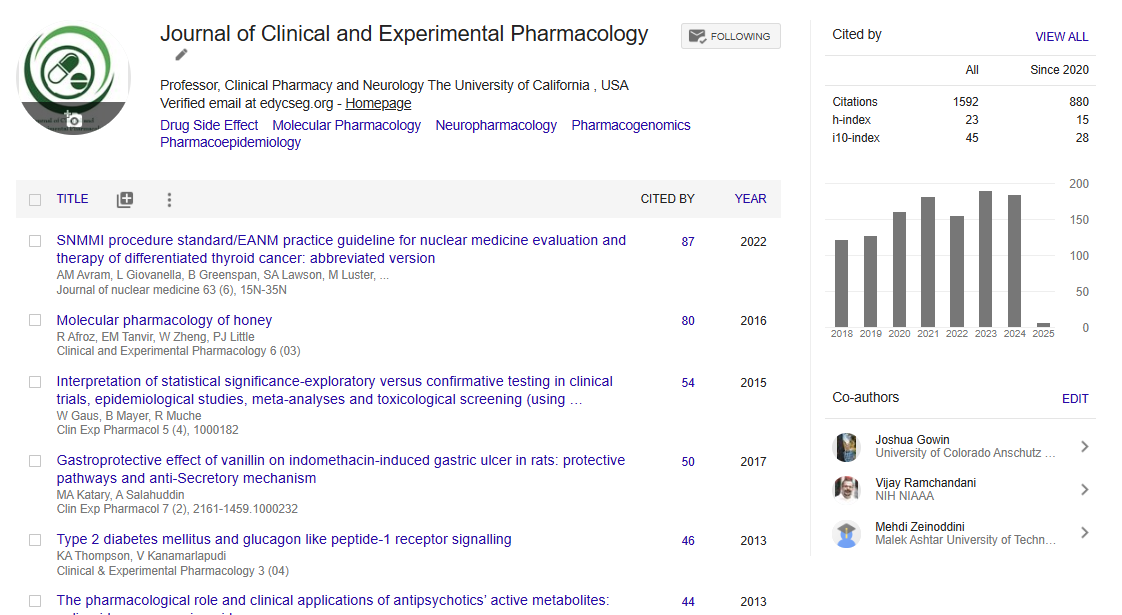

Indexed In
- Open J Gate
- Genamics JournalSeek
- China National Knowledge Infrastructure (CNKI)
- Ulrich's Periodicals Directory
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Opinion Article - (2025) Volume 15, Issue 5
Computational Approaches in Drug Design: Accelerating Pharmacological Discovery
Meera Rao*Received: 02-Sep-2025, Manuscript No. CPECR-25-30502 ; Editor assigned: 05-Sep-2025, Pre QC No. CPECR-25-30502 (PQ); Reviewed: 19-Sep-2025, QC No. CPECR-25-30502 ; Revised: 26-Sep-2025, Manuscript No. CPECR-25-30502 (R); Published: 03-Oct-2025, DOI: 10.35248/2161-1459.25.15.503
Description
Drug discovery is traditionally a time-consuming and resource-intensive process. Conventional methods often involve screening thousands of compounds to identify potential leads, followed by extensive preclinical and clinical testing. In recent decades, computational approaches have emerged as indispensable tools in pharmacology, enabling researchers to predict molecular interactions, optimize chemical structures and identify promising drug candidates before laboratory testing. Computational drug design encompasses a variety of methods, including molecular docking, virtual screening, Quantitative Structure-Activity Relationship (QSAR) Modelling, Molecular Dynamics (MD) simulations and machine learning algorithms. These techniques provide a detailed understanding of drug-target interactions at the atomic level, allowing for the rational design of molecules with optimized efficacy, selectivity and pharmacokinetic properties.
The integration of computational tools with experimental pharmacology has accelerated the discovery of new drugs for complex diseases, including cancer, neurodegenerative disorders, infectious diseases and metabolic syndromes.
Principles of computational drug design
The first step in computational drug design is to acquire accurate information about the biological target. Structural data can be obtained from X-ray crystallography, Nuclear Magnetic Resonance (NMR) spectroscopy, or cryo-electron microscopy. This structural information enables researchers to identify binding pockets, active sites and allosteric regions critical for therapeutic modulation.
Computational drug design can be divided into two primary approaches:
Structure-Based Drug Design (SBDD): Uses the 3D structure of the target to model drug interactions. Molecular docking predicts binding affinities and orientations of potential ligands.
Ligand-Based Drug Design (LBDD): Relies on data from known active molecules to design new compounds, using QSAR models and pharmacophore mapping.
Virtual screening evaluates large chemical libraries in silico to identify compounds with the highest likelihood of binding to the target. This method significantly reduces the number of compounds that need to be synthesized and tested experimentally.
MD simulations model the dynamic behavior of molecules and proteins over time, providing insights into conformational flexibility, binding stability and interaction energies. This information is important for understanding drug efficacy and potential resistance mechanisms.
AI and machine learning algorithms are increasingly applied in drug design to predict bioactivity, toxicity and pharmacokinetics. These models analyze large datasets, uncover hidden patterns and suggest novel molecular structures optimized for therapeutic effect.
Applications of computational drug design
Computational methods have been extensively applied in cancer drug discovery. Kinase inhibitors, Poly(ADP-ribose) Polymerase (PARP) inhibitors and immune checkpoint modulators have been optimized using molecular docking and MD simulations. For example, virtual screening contributed to the identification of novel inhibitors targeting the B-cell lymphoma 2 (BCL-2) family of proteins, which are key regulators of apoptosis in cancer cells.
Drugs targeting Alzheimer’s disease and Parkinson’s disease have benefited from computational design. Structure-based docking has facilitated the development of acetylcholinesterase inhibitors, beta-amyloid aggregation blockers and dopamine receptor modulators, improving both selectivity and efficacy.
Computational approaches accelerate the discovery of antiviral, antibacterial and antifungal agents. During the COVID-19 pandemic, virtual screening of millions of compounds identified potential SARS-CoV-2 main protease inhibitors, demonstrating the rapid application of in silico methods in urgent health crises.
Drug candidates targeting enzymes involved in glucose and lipid metabolism, such as Dipeptidyl Peptidase-4 (DPP-4) inhibitors and 3-Hydroxy-3-Methylglutaryl-Coenzyme A (HMG-CoA) reductase inhibitors, have been optimized using computational techniques to maximize potency while minimizing side effects.
Advantages of computational drug design
Efficiency: Reduces time and cost by limiting the number of compounds requiring synthesis and testing.
Precision: Enables rational design of molecules with high specificity and selectivity.
Prediction: Assesses potential toxicity, bioavailability and resistance mechanisms before experimental studies.
Flexibility: Applicable to multiple disease targets and adaptable to emerging pathogens or novel molecular targets.
Challenges and limitations
Despite its advantages, computational drug design faces several challenges:
Accuracy of models: Predictive models depend on the quality of structural and experimental data; errors in input data can reduce reliability.
Biological complexity: Simulations may not fully capture complex in vivo interactions, including metabolic pathways and off-target effects.
Computational resources: High-resolution Molecular Dynamics (MD) simulations and AI models require significant computational power.
Resistance development: Especially in infectious diseases, pathogens may rapidly evolve, reducing the long-term efficacy of designed drugs.
Future perspectives
The future of computational drug design lies in integrating AI, quantum computing and multi-omics data to create personalized therapies. AI-driven platforms can generate novel drug-like molecules, predict their interactions with multiple targets and optimize their pharmacokinetic and pharmacodynamic profiles.
Integration with systems pharmacology and network medicine will enable the design of multi-target drugs to address complex diseases more effectively. Additionally, advancements in cloud computing and high-performance computing clusters will democratize access to computational tools, allowing smaller laboratories to contribute to drug discovery efforts.
Conclusion
Computational drug design has transformed modern pharmacology, enabling more efficient, precise and cost-effective discovery of therapeutic agents. By integrating molecular modelling, virtual screening, molecular dynamics and artificial intelligence, computational methods allow researchers to predict drug-target interactions, optimize molecular structures and reduce experimental burden. Despite challenges such as biological complexity and computational demands, ongoing technological advancements promise to make computational drug design central to precision medicine and next-generation therapeutics. Its applications in oncology, neurodegenerative disorders, infectious diseases and metabolic conditions underscore its potential to address some of the most pressing medical challenges of our time.
Citation: Rao M (2025). Computational Approaches in Drug Design: Accelerating Pharmacological Discovery. J Clin Exp Pharmacol. 15:503.
Copyright: © 2025 Rao M. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.