
Indexed In
- Academic Keys
- ResearchBible
- CiteFactor
- Access to Global Online Research in Agriculture (AGORA)
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Case Report - (2015) Volume 4, Issue 1
Co-existence of CFTR and SPINK1 Gene Mutations in an Idiopathic Chronic Pancreatitis Case
Abstract
Familial aggregation of CP suggests genetic factors for disease without definitive mode of inheritance. The hypothesized primary putative gene for CP includes SPINK1, CTSB, CTRC, PRSS1 and CFTR. These genes interact with each other and exhibit a variable phenotype in patients. The present report describes a male adult aged 42 years with a complaint of severe recurrent pain in the abdomen and weight loss and the age of onset was 35 years. The family history of chronic pancreatitis was not found. The biochemical examination revealed the exocrine insufficiency. Abdominal CT identified a dilated main pancreatic duct with numerous stones in the pancreas head. Genetic studies identified the patient to be heterozygous for p.N34S and G551D in SPINK1 and CFTR gene. Extended family screening identified his son (10 years) to have the both mutation p.N34S and G551D mutation in heterozygous state. Present findings suggest the need of genetic diagnosis in familial CP cases thereby precaution can be taken to delay or avoid the disease onset.
Introduction
Cystic Fibrosis Transmembrane Regulator (CFTR) gene harbors over 1910 mutations till date (www.genet.sickkids.on.ca). These mutations result in cystic fibrosis or other disease like Congenital Bilateral Absence of Vas Deferens (CBAVD), obstructive azoospermia, bronchiectasis, asthma and Chronic Pancreatitis (CP) etc. [1]. Chronic pancreatitis is a disease of pancreas that is characterized by permanent destruction and fibrosis of the exocrine parenchyma, leading to exocrine pancreatic insufficiency and progressive endocrine failure leading to diabetes.
A familial aggregation nature of CP suggests genetic etiology without definitive mode of inheritance [2,3]. Extensive genetic studies on CP let to classification of hereditary CP and idiopathic CP. Hereditary CP has a penetrance of 70-80% with autosomal dominant inheritance [4]. Idiopathic CP too involves genetic factors but multigenic. The genetic loci reported to predispose CP includes: Serine Protease Inhibitor Kazal 1 (SPINK1), CFTR, CTRC, PRSS1 and cathepsin B (CTSB) [5]. In spite of definitive role of CFTR gene in CP pathogenesis, [6] there are contradictory reports that claim no association of CFTR gene [7,8]. However, recent studies have reported an increased occurrence of CFTR gene mutations in alcohol related CP patients [9,10]. In this case report, we have demonstrated the co-existence of CFTR and SPINK1 gene mutations in an Idiopathic CP cases.
Case Report
A male adult patient aged 42 years visited Gastroenterology OPD of Sanjay Gandhi Postgraduate Institute of Medical Sciences with a complaint of severe recurrent pain in the abdomen and weight loss. A detailed history revealed that the age of onset was 35 years and the patient had no habit of alcohol intake. The family history of CP was not evident. The patient underwent biochemical and radiological examination. Biochemical: Serum amylase test report was normal and pancreatic function test showed only 54.4% (normal value >70%) confirming the exocrine insufficiency. Abdominal CT identified a dilated main pancreatic duct with numerous stones in the pancreas head.
We carried out genetic test of SPINK1, CTSB and CFTR genes. The SPINK1 gene was studied for the most common variants, p.N34S, c.IVS3+2T>C and IVS-37T>C, by PCR RFLP method [11,12]. The CTSB was analysed for p.L26V mutation by PCR RFLP method [13]. The CFTR gene was examined for p.DF508, p.G542X, p.G551D, p.R117H, p.S549N and IVS8 T polymorphism as described by Muthuswamy et al., [10]. The patient was found be heterozygous for p.N34S and G551D in SPINK1 and CFTR gene. Following the mutation identification we explored the mutation status in rest of the family members (Figure 1). We found his son of 10 years old to have both p.N34S and G551D mutations in heterozygous state as in the patient while rest of the family members were mutation free (Table 1).
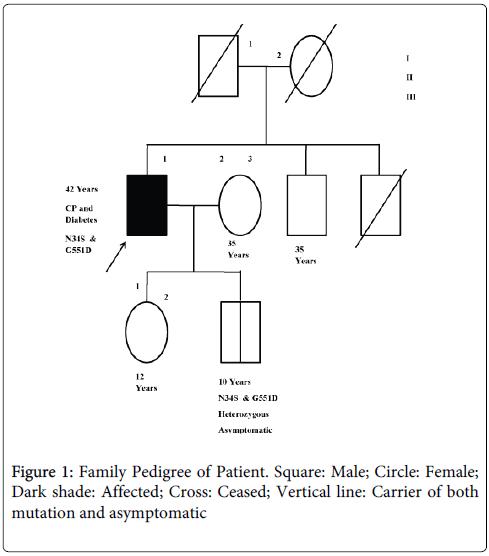
Figure 1: Family Pedigree of Patient. Square: Male; Circle: Female; Dark shade: Affected; Cross: Ceased; Vertical line: Carrier of both mutation and asymptomatic
| Subject | Mutation status | Disease status |
|---|---|---|
| I.1 | Deceased | Uncertain |
| I.2 | Deceased | Uncertain |
| II.1 (Proband) | N34S, G551D carrier | Affected |
| II. 2 | Negative | Unaffected |
| II.3 | Deceased | Uncertain |
| III.1 | Negative | Unaffected |
| III.2 | N34S, G551D carrier | Asymptomatic |
Table 1: Study subjects and their mutation status.
Discussion
In this case report, we reported an idiopathic CP case with heterozygous mutations in SPINK1 and CFTR gene and found one of his son to carry the same mutation. The co-existence of these two mutations supports the multigenic nature of idiopathic CP and interaction of other external factors that could predispose the mutation carrier to CP. Though SPINK1 gene mutations alone have been reported to be present in idiopathic CP, presence of CFTR gene mutation may led to earlier onset of disease or may increase its severity. The presence of CFTR gene mutation in idiopathic CP was reported in few studies [12-14].
The SPINK1 gene encoded protein prevents premature activation of zymogen by trypsin by interacting with trypsin [15]. Polymorphisms of SPINK1 gene result in loss of function and are weakly associated with CP, irrespective of heterozygous or homozygous state. N34S is the most common missense mutation reported in CP. The heterozygous state of this mutation lowers the enzyme level there by premature activation of zymogen results in irreversible damage to pancreases along with CFTR mutation that increases severity further [16].
CFTR channel performs movement of chloride and bicarbonate ions across the pancreatic duct cells there by regulating absorption and secretion of fluids/enzymes. CFTR mutations are hypothesized to result in loss of bicarbonate secretion and cause recurrent acute and chronic pancreatitis rather than cystic fibrosis [8-17]. The reported G551D missense mutation of CFTR falls under class III mutation. These mutations have <1% channel function and display a severe phenotype with pancreatic insufficiency [18] by forming thick mucus in cells. Thick mucus hampers the secreter function of duct cells. The presence of pancreatic exocrine insufficiency in our patient is consistent with the above hypothesis.
Molecular investigation of the family members identified his son as a carrier for both SPINK1 and CFTR mutation. During the study, he was at the age of 10 years and may be a prospective CP patient. The issues have been discussed with the family members about his predisposition for CP.
In conclusion, these data highlight the need of genetic testing in CP patients for effective treatment and family members can be screened to identify any prospective cases they by proper intervention can be started before the disease progresses.
Acknowledgements
Muthuswamy S is thankful to ICMR, New Delhi for fellowship, Singh S is thankful to DST for fellowship and all the authors are thankful for SGPGI, Lucknow for providing infrastructure.
References
- Southern KW (2007)Cystic fibrosis and formesfrustes of CFTR-related disease. Respiration 74: 241-251
- Mohan V, Chari ST, Hitman GA, Suresh S, Madanagopalan N, et al. (1989) Familial aggregation in tropical fibrocalculous pancreatic diabetes. Pancreas 4:690-693.
- Comfort MW, Steinberg AG (1952) Pedigree of a family with hereditary chronic relapsing pancreatitis. Gastroenterology 21: 54-63.
- Lowenfels AB, Maisonneuve P, Dimagno EP, Elitsur Y, Gates LK Jr., et al. (1997) Hereditary pancreatitis and the risk of pancreatic cancer. JNatlCancer Inst 89: 442-446.
- Teich N, Rosendahl J, Toth M, Mossner J, Sahin-Toth M (2006) Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis. Hum Mutat 27: 721-730.
- Shwachman H, Lebenthal E, Khaw KT (1975) Recurrent acute pancreatitis in patients with cystic fibrosis with normal pancreatic enzymes. Pediatrics 55: 86-95.
- Monaghan KG, Jackson CE, KuKuruga DL, Feldman GL (2000) Mutation analysis of the cystic fibrosis and cationic trypsinogen genes in patients with alcohol-related pancreatitis. Am J MedGenet 94: 120-124.
- Sharer N, Schwarz M, Malone G, Howarth A, Painter J, et al. (1998) Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med 339: 645-652.
- Truninger K, Malik N, Ammann RW, Muellhaupt B, Seifert B, et al. (2001) Mutations of the cystic fibrosis gene in patients with chronic pancreatitis.AmJGastroenterol 96: 2657-2661.
- Muthuswamy S, Agarwal S, Awasthi S, Singh S, Dixit P, et al. (2014) Spectrum and distribution of CFTR gene mutations in asthma and chronic pancreatitis cases of North Indian population. Gene 539: 125-131.
- Plendl H, Siebert R, Steinemann D, Grote W (2001) High frequency of the N34S mutation in the SPINK1 gene in chronic pancreatitis detected by a new PCR-RFLP assay. Am J Med Genet 100: 252-253.
- Singh S, Choudhuri G, Agarwal S (2014) Frequency of CFTR, SPINK1, and cathepsin B gene mutation in North Indian population: connections between genetics and clinical data. The Scientific World Journal 2014.
- Mahurkar S, Idris MM, Reddy DN, Bhaskar S, Rao GV, et al. (2006) Association of cathepsin B gene polymorphisms with tropical calcific pancreatitis. Gut 55: 1270-1275.
- Bhatia E, Durie P, Zielenski J, Lam D, Sikora SS, et al. (2000) Mutations in the cystic fibrosis transmembrane regulator gene in patients with tropical calcific pancreatitis. Am J Gastroenterol 95: 3658-3659.
- Bartelt DC, Shapanka R, Greene LJ (1977) The primary structure of the human pancreatic secretory trypsin inhibitor: Amino acid sequence of the reduced S-aminoethylated protein. Arch BiochemBiophys179: 189-199.
- Witt H (2003) Chronic pancreatitis and cystic fibrosis. Gut 52.
- Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, et al. (1998) Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med 339: 653-658.
- Sheppard DN, Welsh MJ (1993) Inhibition of the cystic fibrosis transmembrane conductance regulator by ATP-sensitive K+ channel regulators. Ann NY AcadSci707: 275-284.
Copyright: © 2015 Muthuswamy S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.