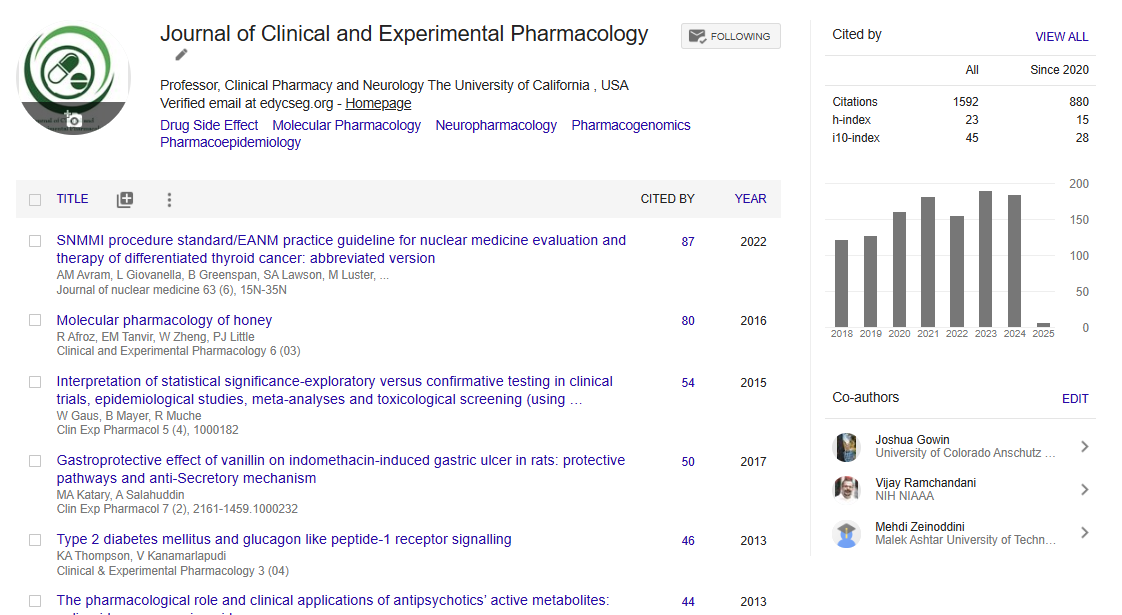

Indexed In
- Open J Gate
- Genamics JournalSeek
- China National Knowledge Infrastructure (CNKI)
- Ulrich's Periodicals Directory
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Review Article - (2024) Volume 14, Issue 1
A Critical Examination of Drug Discovery and its Complex Interplay with the Human Body: A Review
Nishchal Maurya1, Aryan Verma1*, Dheerendra Kumar1* and Ayushi22Department of Biotechnology, Sri Ram Swaroop Memorial University, Lucknow, Uttar Pradesh, India
Received: 08-Mar-2024, Manuscript No. CPECR-24-25107; Editor assigned: 11-Mar-2024, Pre QC No. CPECR-24-25107 (PQ); Reviewed: 25-Mar-2024, QC No. CPECR-24-25107; Revised: 01-Apr-2024, Manuscript No. CPECR-24-25107 (R); Published: 08-Apr-2024, DOI: 10.35248/2161- 1459.24.14.401
Abstract
Drug design is the process of creating novel pharmaceuticals by comprehending a biological target, frequently with the use of bioinformatics and computer modelling techniques. Preclinical research, human clinical trials, and obtaining regulatory clearance for commercialization are all steps in the process.
The creation of new drugs has been expedited by high-throughput screening techniques like Computer-Aided Drug Design (CADD). There are seven phases in the process: Preclinical trial testing, clinical trial testing, pharmacogenomic optimisation, lead compound discovery, lead optimisation, disease selection, and target selection. Clinical trials are FDA-regulated research projects in which participants assist in the exploration for answers to medical problems. Market access, therapeutic effect, monetary rewards, and scientific advancement are all possible outcomes of a successful NDA clearance. In order to manage pharmaceuticals, medical devices, and healthcare goods, guarantee product safety, assess adverse occurrences, and inform regulatory decisions, post-marketing surveillance, or PMS, is essential.
Real-world evidence, patient-reported results, and patient-centric methods are anticipated future trends in regulatory decision-making. Pharmacogenomics is used in personalised medicine to investigate genetic variations and medication interactions; drug-food interactions can impact patient health and result in different treatment outcomes.
Keywords
Biological target; High Throughput Screening (HTS); Lead optimization; Preclinical testing; Clinical trials; Regulatory approvals; Pharmacokinetic interactions; Post-marketing surveillance
Introduction
Drug discovery involves finding new therapeutic entities using clinical, translational, experimental, and computational models. Despite advances in biotechnology, it remains time-consuming, expensive, challenging, and inefficient. Drug design uses computer modelling and bioinformatics to generate innovative medications.
Drug development involves preclinical studies, clinical trials, and obtaining regulatory approval for commercialization [1]. It involves screening hits, medicinal chemistry, and optimization to increase affinity, selectivity, efficacy, metabolic stability, and oral bioavailability. Machine learning techniques have been used to create inhibitors with high activity and target selectivity. Effectiveness is important in drug development to avoid eroded exclusivity agreements and delayed release of me-too medicines.
The success of medications in the commercial and public health sectors depends on a complex interplay of regulators, academia, industry, investors, patent laws, marketing, and communication. The orphan drug funding method ensures patients with rare illnesses are unlikely to benefit from pharmacotherapeutic advancements. Nonsteroidal anti-inflammatory drugs are commonly used, but they have risks. Gouda AM and colleagues developed and tested N-(4- bromophenyl)-7-cyano-6-substituted-H-pyrrolizine-5-carboxamide derivatives as dual COX/5-LOX inhibitors [2]. These compounds could serve as a foundation for future research into safer gastric properties.
Literature Review
Drug discovery process
The average cycle lasts about 14 years, costing between 0.8 and 1.0 billion dollars. However, recent breakthroughs in combinatorial chemistry and high-throughput screening methods have sped up the process by allowing vast libraries of compounds to be screened and synthesized quickly. As a result, numerous approaches have been developed to accelerate the research cycle while lowering expenses and the likelihood of failure in drug discovery. Computer-Aided Drug Design (CADD) is one of the most effective ways to achieve these goals.
CADD refers to the computational tools and resources used to store, organize, analyse, and model compounds [3]. It addresses a broad spectrum of drug development topics, including compound synthesis software, tools for methodically assessing possible lead candidates, and the production of digital libraries for investigating chemical interactions. From target identification and validation to lead discovery and optimization; the tools can even be used in preclinical trials, dramatically changing the drug discovery and advancement process.
Common computational drug discovery strategies include Structure-Based Drug Design (SBDD), Ligand-Based Drug Design (LBDD), and sequence-based approaches. SBDD uses crystal structures, NMR data, and homology models to understand target macromolecule structure. LBDD uses tools like QSAR for drug target-ligand interactions [4]. Sequence-based approaches use bioinformatics tools to identify targets early and conduct lead discovery in drug discovery. Combinational and hierarchical methods influenced by technological factors like conformation generation, sampling, scoring systems, optimization algorithms, and chemical similarity calculations, are widely used.
The drug development process is divided into seven steps: Disease selection, target selection, lead compound identification, lead optimisation, preclinical trial testing, clinical trial testing, and pharmacogenomic optimisation. To improve efficiency, efforts focus on ligand discovery and optimization, with the weakest link being the discovery of new targets.
Target identification and validation
The process of finding tiny compounds that can have a strong and selective effect on the functions of target proteins. Natural compounds have historically been abundant, and knowledge of their lethal or therapeutic effects frequently predates comprehension of their particular target or mechanism [5]. In recent years, efforts to find novel small-molecule probes and therapies have switched to screening a diverse variety of compounds to select those that elicit the desired biological response. Target-based screenings use in vitro biochemical assays to identify compounds interacting with molecular entities in diseases. Phenotypic screens, on the other hand, use cell-level assays to determine desirable phenotypic changes.
The most popular phenotypic test is the high-content assay, which uses imaging technologies to track the effects of drugs on developing cells, sometimes with cell-based readouts. It’s significance noting that drug development began using phenotypic screens before target-based screens became prominent in the 1980s. To put it simply, the process of generating innovative drugs entails discovering small compounds capable of potently and selectively altering the activities of target proteins. Endpoint assays determine molecule activity per well, accelerating the analogue development phase by developing early screens with a specific molecular target [6].
Trastuzumab and its variations, a first-in-class immunotherapeutic that targets the HER2 tyrosine kinase receptor and is used to treat patients with HER2-overexpressing breast tumours, are an excellent illustration of the role of TID in personalised medicine. Within a year of infection, the combo therapy stopped HIV-1 infection from developing to a fatal stage.
The target-based technique, however, has a significant drawback in that success necessitates a thorough comprehend of the disease’s aetiology. Prior information existed in the cases of HIV-1 and breast cancer, resulting in a logical TID/MoA for therapeutic development. Inadequate preclinical target validation-that is, gaining clear proof that the target chosen is closely connected to the condition of interest and should have therapeutic value-has been a primary cause of medications found via target-based techniques failing in clinical trials.
Many complicated illnesses, particularly brain disorders, have sporadic forms or are difficult to identify essential molecular targets, leaving just a few plausible molecular failure scenarios [7]. According to a recent study, CRISPR-based genetic knockouts of six distinct protein targets for ten different anti-cancer medications did not hinder the treatments’ ability to kill cancer cells, suggesting that the initial targets were incorrect (Figures 1 and 2).

Figure 1: A flow chart in above figure illustrates the methodical procedure for target identification and validation in drug discovery.

Figure 2: The two primary methods for carrying out the procedure are (A) Phenotype-Based (forward chemical genetics) and (B) Target-Based (reverse chemical genetics).
Hit generation
High-Throughput Screening (HTS) is a method used to identify new pharmaceuticals by screening and analysing biological modulators and effectors against specified targets [8]. It is employed in various fields, including peptide, protein, and combinatorial chemical libraries. HTS aims to accelerate drug detection by screening large chemical libraries at rates of over a few thousand compounds per day or week.
Frozen or dried samples of compounds to be screened are placed on microtiter plates in the library for testing. HTS research and development have a wide range of applications, including enzyme testing, whole organ testing, and whole animal testing with cassette dosage. HTS can also improve current drug moieties and increase their efficacy. As more human genomes are added, the chemical library might have up to 100 million options.
There are two types of tests: Homogenous and heterogeneous. Homogeneous tests are simpler and less expensive, while heterogeneous assays are more demanding. Fragment-based ligand discovery is a great technique for creating high affinity ligands with the necessary physiochemical qualities to serve as therapeutic leads [9]. The drug discovery hit generation process is divided into several steps: Target selection, target validation, assay development, compound library screening, primary screening, hit confirmation, hit characterization, hit optimization, lead identification, preclinical testing, and lead compound screening.
Hit production is important early in the drug discovery process, as finding successful hits facilitates the subsequent phases of development and optimization. Hit discovery and confirmation are important stages in drug development, involving chemical screening assays and retesting to confirm a hit. Assays can be biochemical or cell-based, and pharmacological significance, repeatability, quality, and chemical repercussions must be considered. Drug-like compounds undergo in vitro tests, dose-response curves, secondary assay analysis, and basic structure-activity relationship data [10].
Lead optimization
Lead optimization is important for pharmaceutical firms to accelerate research and reduce costs by evaluating treatment leads’ early develop ability and ADMET features, addressing potential compound failures [11].
Inadequate physicochemical characteristics can lead to questionable findings and negative impacts on overall costs and clinical outcomes. It is important to optimize drug-like characteristics and pharmacological potency against the target. A prospective lead drug with strong in vitro receptor affinity and selectivity should not be discarded due to insufficient physicochemical characteristics. In silico computation of pKa, solubility, and lipophilicity is essential for library synthesis planning. Lead optimization focuses on creating preclinical candidates from “Hit” or “Lead,” increasing efficacy in target potency, selectivity, protein binding, and pharmacokinetic features. New drug molecules often fail due to issues like unreliable assays, unsuitable targets, potential toxicity, unfavourable side effects, and poor PK/PD profiles. The development process involves modelling, data testing, and analysis [12]. A successful drug molecule should have few adverse effects and be effective in a specific pathway leading to its biological target. Identifying and confirming the target interest is important for successful drug discovery for specific illnesses.
The manufacture of protein function modulators requires biological targets, including genes, proteins, RNA, and DNA [13]. Among the target identification techniques are small-interfering RNAs (siRNAs), phenotypic screening, mRNA/protein level analysis, genome-wide forward and reverse genetics screening, gene expression monitoring, and genetic linkages (Figure 3).

Figure 3: Lead optimization is an important step in drug discovery, enhancing efficacy and target potency, preventing failures due to safety concerns, inadequate research, and toxicity.
Pre-clinical testing
Preclinical research is important for evaluating drug or method viability before clinical trials. However, inadequate planning and execution can lead to resource squandering and experimental biases [14]. Preclinical research, categorized into confirmatory and exploratory research, focuses on evaluating theories and developing hypotheses about disease pathophysiology. It requires adaptability and rigor in planning, execution, evaluation, and publication to demonstrate repeatable intervention effects in animal models [15].
The development of pre-clinical drugs involves various life sciences specialties, including target discovery, compound screening, biological validation, mechanistic characterization, and optimization. The pharmaceutical industry faces challenges due to 90% of medical discoveries never becoming commercial products. European integration allows local experts to organize large-scale research projects in advanced pharmaceutical chemistry, placing a significant responsibility on life scientists [16]. Medicinal chemistry aims to develop a novel chemical entity that interacts systemically with the target, demonstrating potency, selectivity, specificity, and overall patient safety.
The development of innovative drugs requires strict non- pharmacological requirements, including well-specified intellectual property rights, a technically feasible, affordable, and easy-to-scale process, and high chemical purity of the Active Pharmaceutical Ingredients (API). Pharmaceutical legislation and regulations are being enforced by major markets like the US and Japan. Preclinical testing is important for assessing safety, efficacy, and pharmacokinetics in animal models [17]. Three study approaches are used: Ex vivo, in vivo, and in vitro experiments. Poor design and implementation can hinder clinical translation and waste resources. Researchers can reduce experimental biases and improve scientific rigor by following general design guidelines for hypothesis testing investigations (Figure 4).

Figure 4: The figure describes the pre-clinical research phase of drug development, which includes identifying a target, conducting pharmacokinetic and pharmacodynamic tests, toxicology studies, dose optimization, and in vitro and in vivo testing. Success is documented in an IND application, ensuring safety and ethical compliance.
Investigational New Drug (IND) application
The FDA requires an Investigational New Drug (IND) application before human drug testing, ensuring safety, effectiveness, and toxicity through USP-based research.
The FDA’s section 312 outlines three conditions for exempting a drug from IND requirements: The drug is legally sold in the US, the marketed product’s medicine has not undergone significant changes, there is not much evidence that prescription drugs change the need for advertising, the study excludes patient demographics, method of administration, dose, and a complete review before beginning the study [18]. For an IND application, three conditions must be satisfied, the investigator uses it in the patient population and dose form indicated on the label, permitting the use of the experimental drug raises or lowers the risk, and a well-controlled commercial drug intended to have a major impact on labelling or promotion. Form 1571 covers all applications, except for sponsor- investigators [19]. Clinical studies must be submitted on various forms, including Form 1572 and Form 3674, and must provide toxicological and chemical information, reports of irregular or failed clinical investigations, and safety reports.
Clinical investigators in pharmaceutical trials often follow regulatory requirements, such as filing an Investigational New Drug (IND) application if research meets exemption standards. If an individual investigator meets FDA qualifications, the application process is less complex [20]. Three FDA forms are required: FDA Form 1571, 1572, and 3674. If approved, trials can start 30 days later. However, for lone investigators unfamiliar with regulatory procedures, filing an IND can be intimidating and a barrier to conducting pharmacological research, particularly when starting a drug study (Figure 5).

Figure 5: The Investigational New Drug (IND) application is an important step in the drug development process. Here are the general steps involved in filing an IND application with regulatory authorities, such as the U.S. Food and Drug Administration (FDA).
Clinical development
Phase I clinical trials: Phase 1 clinical trials are important in drug development, aiming to determine a new medicine’s safety and tolerability in humans. These trials involve a small group of healthy volunteers, often 20 to 100, who undergo a dose-escalation design to understand the drug’s behavior, safety profile, metabolism, and excretion. The data is then analyzed to determine the drug’s safety profile, potential adverse effects, and the best dose for Phase 2 and Phase 3 studies [21].
Phase II clinical trials: Phase 2 of a clinical trial is an important phase in drug development, testing a medicine on a larger group of participants to determine its safety and effectiveness. Participants are carefully chosen based on factors like illness type, past therapies, and health state. The efficacy assessment measures the drug’s effectiveness in treating the specific disease. Phase 2 data is used for dose optimization and regulatory assessment to determine if the medicine can proceed to Phase 3 trials [22].
Phase III clinical trials: Phase 3 clinical trials are important in developing new medicines or therapies, aiming to determine their effectiveness and safety compared to existing treatments. These trials involve a larger number of participants and are conducted in various locations [23]. The trials are designed as randomized controlled trials, with participants randomly assigned to receive the new medication, usual treatment, or a placebo. Data collection involves collecting health outcomes and statistical analysis to determine the effectiveness and safety of the treatment [24].
New Drug Application (NDA) submission
A New Drug Application (NDA) is a formal request for permission to market a new pharmaceutical product, requiring extensive data on development, safety, effectiveness, manufacture, and labelling to regulatory bodies [25]. An NDA submission includes clinical and nonclinical pharmacology, pharmacokinetics, drug interactions, patient demographics, study protocols, and more. It includes bio- statistical techniques, data summaries, pediatric studies, and risk management techniques. The European Medicines Agency and FDA review NDAs, with the CHMP reviewing a single application.
Pharmaceutical companies must submit National Drug Applications (NDAs) to various regulatory bodies worldwide for drugs intended for international markets. The process involves pre-submission planning, data preparation, document compilation, and quality assurance. Many regulatory bodies now demand electronic submissions, and a filing fee is required to cover agency evaluation expenses [26]. The first agency review includes acceptance review, requests for additional information, and advisory committee meetings. If accepted, the regulatory body issues an approval letter, allowing the company to market and sell the medication. A successful NDA can increase a pharmaceutical company’s market value, attract investors, and foster collaborations. Upcoming trends in the NDA process include global harmonization, artificial intelligence, faster-track designations, priority review, real-world evidence, regulatory convergence, patient-centric approaches, and digital transformation [27].
Assembling all information from clinical and preclinical studies in order to receive regulatory approval: The complexities of data compilation in drug development, focusing on essential elements, regulatory requirements, and obstacles faced by pharmaceutical companies, highlight its important role in launching new therapeutic entities [28].
Pharmacology studies examine medication interactions with biological systems, target receptors, and efficacy. Animal research, molecular assays, and in vitro experiments determine drug characteristics. Toxicology studies evaluate safety, while Phase II, I, and III trials assess effectiveness in diverse patient populations. Special population studies provide additional information [29].
Regulatory agencies like the European Medicines Agency (EMA), the Committee for Medicinal Products for Human Use (CHMP), and the U.S. Food and Drug Administration (FDA) set strict guidelines for compiling data for drug approval. The Common Technical Document (CTD) is an internationally recognized format for compiling and presenting data. Technological advancements like cloud-based platforms and data analytics tools can help increase efficiency. Data compilation is essential for proving a drug’s safety, ensuring patient safety, and improving industry success [30]. Future trends in data compilation include the incorporation of machine learning and artificial intelligence, real-time data monitoring, patient-centric strategies, and international cooperation. These developments will facilitate data compilation and enhance the overall effectiveness of drug development [31].
Regulatory approval: A government regulatory agency’s formal clearance or authorization to permit the marketing, sale, or use of a novel medication, medical device, therapeutic intervention, or other healthcare product is referred to as regulatory approval [32]. Before these products are made available to the general public, the regulatory approval process is an important step in ensuring that they meet established standards for quality, safety, and efficacy. Regulatory bodies are essential in assessing the information provided by producers or developers and in deciding which healthcare products to approve, label, and conduct post-market surveillance [33].
Different regulatory bodies are in charge of monitoring approvals in their respective jurisdictions, and the regulatory approval procedure differs between nations and regions. The Therapeutic Goods Administration (TGA) in Australia, the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan, the European Medicines Agency (EMA) in the European Union, and the U.S. Food and Drug Administration (FDA) in the United States are a few prominent regulatory bodies [34].
Review and approval
Drug-Drug Interactions (DDIs) can lead to increased toxicity or decreased therapeutic efficacy, affecting patient safety and therapeutic outcomes. FDA guidelines use in vivo total plasma and blood concentrations and in vitro Ki values to assess DDI potential during drug development and regulatory review [35].
Since the mid-1990s, the FDA has been using in vitro models to guide future in vivo studies on pharmacokinetic drug delivery inhibitors (DDIs). These documents offer recommendations for dosage adjustments and cautionary language in labels. The FDA’s guidance documents are not individual rights but reflect its current stance on drug interactions. Understanding the mechanisms underlying DDIs is important for patient safety and therapeutic outcomes (Figure 6) [36].

Figure 6: A New Drug Application (NDA) is a request for permission to market a new pharmaceutical product, approved by the FDA and EMA in the US and EU. Successful NDAs boost market value, attract investors, and foster collaborations.
Regulatory agencies review the NDA for safety, efficacy, and quality: A pharmaceutical company submits a New Drug Application (NDA) to a regulatory body for approval to market and sell a new drug. The NDA review process includes data compilation, screening, acceptance review, and multidisciplinary review teams. The Common Technical Document (CTD) structure organizes data, and initial screening ensures submission meets standards [37]. The efficacy of a drug is determined through patient subgroup analyses, confirmatory evidence, and consistency across trials, and clinical trial data analysis. Quality assurance is ensured through inspections and stability tests. Regulatory agencies may request an advisory committee review, providing unbiased opinions and suggestions.
Manufacturers and healthcare professionals must report any negative side effects or safety issues, and stakeholders should be informed about new safety information. Review teams assess data, including preclinical safety data, clinical safety data, adverse event reporting, benefit-risk analysis, and toxicology studies, to identify potential risks and suggest mitigation techniques [38]. The NDA review process ensures the security, effectiveness, and quality of new medications (Figure 7).

Figure 7: The NDA submission process involves a thorough evaluation of a drug’s effectiveness and safety, adherence to Good Manufacturing Practices, patient subgroup analyses, clinical trial data analysis, and post-market surveillance, Advisory Committee Review, and risk mitigation techniques.
Approval or rejection of the drug for marketing: The regulatory pathway involves approving or rejecting a drug for marketing based on its quality, safety, and efficacy, involving a comprehensive benefit-risk assessment, therapeutic benefits-risks weighing, and multidisciplinary teams [39].
The drug approval process involves analysing clinical and preclinical studies, manufacturing and quality control data, and scientific evidence to make decisions. The NDA’s strength is the foundation for evidence-based approval, which requires proven efficacy, clinical significance, favourable benefit-risk profile, patient-centred approach, strict manufacturing standards, batch consistency, regulatory compliance, openness, and accountability [40]. The decision to approve or reject a medication impacts patient access, public health, and the pharmaceutical sector.
Regulatory flexibility in pharmaceuticals allows for accelerated approval pathways and the designation of drugs as orphan drugs for rare diseases, ensuring faster access to medications. However, rejections can result from inadequate efficacy data, safety concerns, or manufacturing issues [41]. Complete Response Letters (CRLs) help pharmaceutical companies address these issues and resubmit the NDA. Public health impact is considered in regulatory decisions, affecting overall healthcare outcomes. Clear communication promotes public trust and control expectations. Successful approvals improve a company’s market standing, but development strategies may need to be reassessed in case of rejection. The approval process is important for patient safety and effectiveness. Predictive modelling and advanced technologies like artificial intelligence and machine learning will aid decision-making. Global regulatory standardization aims for uniform and effective reviews across jurisdictions [42].
Post-marketing surveillance
A critical component of the lifecycle management of medications, medical equipment, and other healthcare products is post-marketing surveillance, or PMS. It entails the methodical observation and assessment of these products’ efficacy and safety subsequent to their approval and release onto the market. In order to guarantee the continued safety of products, evaluate adverse events, and guide regulatory decisions, PMS is important [43].
PMS aims to detect adverse events, evaluate product effectiveness, and balance risks and benefits. Early detection identifies unanticipated hazards or safety issues not fully recognized in pre- market trials. Real-world effectiveness considers patient adherence and comorbidities. Risk-benefit analysis helps patients, healthcare providers, and regulators make informed decisions about product use. Long-term safety evaluations ensure product quality by evaluating cumulative effects, detecting gradual adverse events, and monitoring manufacturing procedures [44].
Post-marketing surveillance involves various techniques like data mining, registries, observational studies, statistical methods, spontaneous reporting systems, and signal detection and analysis. These techniques work with healthcare systems to access patient data in real time for on-going observation. Regulatory frameworks for post-marketing surveillance include international collaboration with ICH, regional regulations like FDA and EMA, risk evaluation and mitigation strategies, post-approval commitments, and communication systems [45]. Real-world data will be used for on-going safety monitoring, with patient participation being key. Predictive analytics and other technologies will be used to identify safety issues early. Harmonization efforts and shared databases will be part of global collaboration for effective data exchange among regulatory agencies [46].
Continuous monitoring of the drug’s safety and effectiveness after it enters the market: Pharmacovigilance is the continuous assessment of a product’s performance in real-world settings, identifying potential safety issues not discovered during pre- market clinical trials. It involves monitoring a drug’s safety and effectiveness post-market, gathering, evaluating, and interpreting adverse events information, and assessing its efficacy across various patient populations [47].
Continuous monitoring is important for identifying hidden risks, revealing uncommon effects, evaluating effectiveness in real-world settings, comparing effectiveness to other treatments, validating results from clinical trials, and assessing long-term advantages. It is essential for ensuring the safety of pharmaceutical products and enables a dynamic re-evaluation of the risk-benefit ratio. Continuous monitoring methodologies include spontaneous reporting, data mining, observational studies, predictive modelling, comparative effectiveness research, and advanced analytics [48]. However, challenges include underreporting, data integration, quality, signal specificity, resource limitations, and global variability. To overcome these, efficient channels for transparency and communication are essential for on-going monitoring efforts.
Ensuring drug safety and efficacy requires open communication, cooperation between regulatory bodies and the pharmaceutical industry, and real-world data analysis. Adopting new technologies, patient involvement, international cooperation, and incorporating real-world data into decision-making are important for continuous monitoring [49]. As healthcare systems evolve, embracing technological advancements, integrating real-world evidence, involving patients in monitoring, and fostering global collaboration are essential. Advanced technologies like AI and machine learning are being integrated for effective signal detection and data analysis, enabling safe and efficient care worldwide [50].
Drug interaction
Drug interactions occur when substances like meals, drinks, or medications are consumed simultaneously, altering a drug’s mechanism of action. Examples include grapefruit’s impact on pharmaceutical metabolism and the simultaneous targeting of receptors, such as alcohol and zolpidem [51]. These interactions can lead to additive, synergistic, or antagonistic effects. Different medications have different effects, making it difficult to differentiate between them. Direct interactions between medications, such as thiopentone and suxamethonium, can occur when combined before intravenous administration. However, the individual impacts of these interactions can be difficult to distinguish (Figure 8) [52].

Figure 8: Drug interaction in pharmaceutical sciences involves substances altering a drug’s mechanism of action, such as grapefruit interactions. It involves receptor targeting, drug-drug interactions, and direct interactions including Pharmacokinetics and Pharmacodynamics Interactions. Identifying and distinguishing between these interactions is challenging.
Pharmacokinetic interactions
The area of pharmacology known as Pharmacokinetics (PK) is dedicated to the study of drug metabolism in the human body. It deals with drug absorption, distribution, metabolism, and excretion; these processes are often abbreviated as ADME. Comprehending pharmacokinetics is essential for ascertaining the drug’s concentration at the site of action over an extended period and forecasting its physiological effects [53].
How the body absorbs, distributes, metabolizes, and excretes drugs:
A) Adsorption: Absorption is the process by which a medication enters the bloodstream. Common delivery methods include oral, intravenous, intramuscular, subcutaneous, and transdermal. Oral absorption is popular due to its ease of administration through the gastrointestinal tract. Intravenous absorption allows for rapid and complete drug absorption [54]. Intramuscular and subcutaneous administrations compromise speed and convenience. Transdermal absorption is suitable for prolonged therapeutic levels. Factors such as drug physicochemical characteristics, formulation, gastrointestinal motility, and potential interactions with food or other medications affect drug absorption [55].
B) Distribution: Drug distribution involves entering the bloodstream and spreading throughout the body, influenced by factors like drug affinity, tissue permeability, and blood flow. The binding of plasma proteins, particularly albumin, affects drug distribution.
The Blood-Brain Barrier (BBB) limits drug flow into the brain, impacting central nervous system distribution [56]. Pregnant women may breach the BBB, affecting fetus safety. Tissue perfusion affects drug distribution, with highly perfused organs absorbing drugs more easily. The volume of distribution (Vd) is a pharmacokinetic measure that reveals drug distribution.
C) Metabolism (biotransformation): The body converts medications into metabolites through metabolism, with the liver being the main organ involved. The cytochrome P450 (CYP) enzyme system is responsible for enzymatic reactions. Phase I metabolism introduces functional groups through oxidation, reduction, and hydrolysis [57]. Phase II metabolism, or conjugation, attaches water-soluble molecules to the drug. Enzyme induction and inhibition occur at the metabolic level, where a medication affects the function of metabolizing enzymes. Induction can increase drug metabolism, reduce efficacy, while inhibition can decrease clearance and increase toxicity risk [58].
D) Excretion: Drugs and their metabolites are excreted through various pathways, including the kidneys, liver, and breath. Kidneys filter drugs based on size, charge, and solubility, while the liver excretes them as bile [59]. Some drugs are easier to excrete through active transport or passive diffusion. Sweat and breath are common methods, but they make up little of the total excretion. Clearance is important for drug dosing regimens.
Pharmacokinetics, a field of study that involves understanding and predicting drug behavior using mathematical equations and models, is important for maximizing drug therapy in clinical settings [60]. Key parameters include AUC, bioavailability, and half-life. Understanding pharmacokinetics is especially important for special populations like juvenile and geriatric patients, pregnant women, and those with compromised organ function. Doctors must also carefully examine potential drug-drug interactions to ensure effective treatment and minimize side effects (Figure 9) [61].

Figure 9: The figure illustrates Pharmacokinetics, the study of drug metabolism, including absorption, distribution, metabolism, and excretion. It covers various methods, challenges in oral absorption, distribution, metabolism, and excretion, and the Blood-Brain Barrier. This is important for optimal therapy, dosage adjustments, and drugdrug interactions.
Pharmacodynamic interactions
Pharmacodynamics is a field of pharmacology that studies how medications affect the body and the processes that lead to these effects. It focuses on how a medicine interacts with its intended target, such as a receptor, enzyme, or ion channel [62]. The main goals of pharmacodynamics are to understand how pharmaceuticals exert their therapeutic or harmful effects and the connection between drug concentration and response. Drug interactions with receptors, such as nuclear, ion channel, G protein-coupled and enzyme-linked receptors, are studied. The dose-response relationship is important for determining the right dosage to optimize therapeutic effects and minimize toxicity and side effects [63]. The therapeutic index, determined by dividing the minimal hazardous dose by the minimum effective dose, serves as a gauge for a medication’s safety margin. A larger safety margin indicates a lower probability of side effects. Prolonged exposure to certain medications can lead to desensitization and tolerance, necessitating higher dosages [64].
How drugs affect the body’s functions: Pharmacodynamics studies drug interactions with proteins or macromolecules to trigger a response. Drug-receptor interactions are important, with receptors like GPCRs, enzyme-linked receptors, nuclear receptors, and ligand-gated ion channels playing a role [65]. Antagonists mimic receptor activation, inhibiting agonists or endogenous ligands. The equilibrium between agonists and antagonists determines the overall pharmacological action.
GPCRs, including opioid and beta-adrenergic receptors, are involved in physiological processes. Nuclear receptors, also known as transcription factors, control gene expression and protein synthesis [66]. Steroid hormone receptors and peroxisome proliferator- activated receptors are two types of nuclear receptors. Drug- receptor interactions are often associated with second messenger systems like cAMP, cGMP, and IP3, which transmit signals from the cell membrane to intracellular compartments. Protein stability, location, and activity can be altered during signal transduction. Transcription factors regulate gene transcription, and their levels can be impacted by drug-receptor interactions [67].
Drug-receptor interactions are linked to second messenger systems, such as cAMP, cGMP, and IP3, which transmit signals from the cell membrane to intracellular compartments. These interactions can affect protein stability, location, and activity, as well as transcription factors. The endocrine, immunological, pulmonary, gastrointestinal, and central nervous systems all impact a drug’s physiological response and therapeutic effects. Genetics, environment, and medical problems also influence an individual’s medication reaction. Personalized medicine relies on pharmacogenomics to understand genetic variations in drug-metabolizing enzymes and receptors [68]. Adverse Drug Reactions (ADRs) can still occur, and drug resistance and tolerance pose significant challenges in treating infectious illnesses and cancer.
Synergistic or antagonistic effects between drugs: Synergy refers to the collaboration between two or more medications, resulting in a larger outcome than the sum of their separate effects. This can be achieved through receptor activation, complimentary activities, pharmacokinetic interactions, and the combination of medications with various modes of action. Complementary activities involve altering enzymes, transporters, or cellular processes, while variations in medication absorption, distribution, metabolism, or excretion can impact synergistic effects [69]. Clinical examples of synergy include analgesic combinations, chemotherapy for cancer, and antibiotic combinations in infectious disorders. Dosage modifications are essential to avoid toxicity. Antagonistic effects in pharmacodynamics involve the interaction between two or more drugs, resulting in reduced overall effects compared to each drug alone [70]. Mechanisms of antagonism include competitive receptor blockade, functional antagonism, and pharmacokinetic interactions.
Antagonism is a clinical phenomenon where drugs interact with each other, causing agonists in some situations and antagonists in others. Examples include beta-blockers and beta-agonists for hypertension and asthma, and naloxone in overdose treatment. Combining anticoagulant drugs with antiplatelet agents can lead to antagonistic effects, as anticoagulants inhibit blood clotting while antiplatelet agents prevent platelet aggregation. Dose adjustments may be necessary to overcome these effects. Regular clinical monitoring is important for guiding treatment plan adjustments and identifying signs of inadequate therapeutic response or increased side effects. Understanding these temporal facets is essential for effective therapeutic administration [71].
Drug-drug interactions
The effects of taking two or more drugs at the same time that change their pharmacological activity are known as drug-drug interactions, or DDIs. Increased toxicity or decreased therapeutic efficacy may result from these interactions. Healthcare workers must comprehend the types, mechanisms, and clinical importance of DRIs in order to guarantee safe and efficient medication [72]. DRIs can appear during various phases of drug metabolism and action, including metabolism, conjugation reactions, distribution, absorption, and elimination.
Drug absorption and distribution can be influenced by factors such as tissue binding, protein binding, altered stomach pH, and competition for absorption sites in the gastrointestinal tract. Drugs may also compete for conjugation enzymes in conjugation reactions, leading to interactions [73]. Elimination can occur through renal clearance mechanisms, altered excretion rates, and inhibition of transporters. Understanding these interactions is important for healthcare professionals to ensure safe and effective pharmacotherapy. Drug-drug interactions can lead to enhanced toxicity, modified pharmacokinetics, drug level fluctuations, and therapeutic failure. Age-related variations in drug metabolism and clearance may make certain populations more vulnerable to drug interactions. Polypharmacy, the use of multiple medications, increases the risk of drug interactions. Warfarin can be displaced from protein binding sites, increasing bleeding risk. Azole antifungals can inhibit cytochrome P450 enzymes, increasing statin metabolism risk [74].
Combined effects of two or more drugs taken simultaneously: Polypharmacy, or drug combinations, involves the use of multiple medications to enhance therapeutic efficacy or minimize adverse effects. Healthcare workers must understand the rules governing these combinations to ensure patient safety and maximize treatment results. Combinations can treat complex conditions like diabetes or hypertension, reduce side effects, and broaden coverage against bacterial infections [75]. They can also delay drug resistance in microbial infections, particularly in HIV, TB, and some cancers.
Pharmacokinetic and pharmacodynamic interactions influence drug combinations, with dose-response relationships important for forecasting the overall effects. Examples of therapeutic drug combinations include antiretroviral therapy for HIV, combination chemotherapy for cancer treatment, antihypertensive therapy, and antibiotic combinations for specific bacterial infections. However, these combinations come with risks, such as polypharmacy in the elderly, complicated dosing schedules, and increased side effects [76]. Healthcare providers should use evidence-based guidelines, conduct regular medication reviews, educate patients about prescribed regimens, and customize treatment plans based on unique patient characteristics to optimize drug combinations. Consistent observation and tracking can help identify and control negative consequences or issues.
Potential for altered therapeutic effects or increased toxicity: Personalized medicine is a future direction in drug combinations, using technology like clinical decision support systems and electronic health records to detect possible drug interactions and improve treatment plans. Pharmacogenomics advances could result in more individualized drug combinations that take into account a person’s metabolism and genetic composition. Drug administration can result in increased toxicity or altered therapeutic effects due to factors such as drug interactions, patient-specific characteristics, and pharmacokinetics and pharmacodynamics [77].
Pharmacokinetic factors include bioavailability, competition for absorption sites, gastrointestinal motility, protein binding, and tissue penetration. Factors like food, gastric pH, and other factors can affect bioavailability, which can lessen the overall therapeutic effect. Protein binding can affect the concentration of a free drug in the bloodstream, potentially leading to sub-therapeutic or toxic effects. Tissue penetration is influenced by factors like tissue perfusion and drug lipophilicity, which can alter drug distribution into tissues and impact therapeutic outcomes.
Receptor interactions, functional selectivity, and enzyme induction or inhibition are also important pharmacodynamic factors. Factors like lithium and NSAIDs, digoxin and diuretics, warfarin and antibiotics, and patient education can all impact drug-drug interactions. To avoid bleeding complications, closely monitor the International Normalized Ratio (INR) and make necessary dosage adjustments.
By addressing these factors, healthcare workers can ensure the best possible therapeutic results and minimize side effects [78].
Drug-food interactions
Drug-food interactions are important in healthcare to help patients maintain a healthy, balanced diet and maximize their prescription regimens. These interactions can occur through competition for absorption sites, the impact of food on stomach emptying, metabolic interactions, and nutrient-drug interactions [79]. Drug bioavailability can be impacted by absorption interactions, which involve contests for absorption sites in the gastrointestinal tract. Foods can affect the liver’s cytochrome P450 enzyme activity, which is involved in metabolic interactions. Compounds that inhibit cytochrome P450 3A4 are involved in nutrient-drug interactions, which can result in higher drug concentrations and possible toxicity. Protein binding in the bloodstream during distribution interactions can alter the concentration of drugs that are not bound and may result in altered therapeutic effects or unfavourable reactions.
Drug-food interactions can lead to reduced efficacy, increased efficacy, toxicity, and nutrient imbalance. Patients may need to adhere to specific dietary restrictions, which can affect their willingness or ability to comply with treatment regimens [80]. Genetic factors can also influence individual responses to drug- food interactions, and understanding a patient’s genetic profile can help predict their susceptibility to specific interactions. Drug- food interactions can have serious clinical ramifications, such as warfarin and vitamin K, levothyroxine and calcium supplements, tyramine-containing foods, and Monoamine Oxidase Inhibitors (MAOIs). Warfarin users should continue to consume these foods consistently and let their doctor know about any major changes. Patients taking Monoamine Oxidase Inhibitors (MAOIs) may experience hypertension crises due to the MAOIs’ ability to prevent the breakdown of tyramine, a substance present in some foods. Patients should be informed about dietary restrictions, when to take their medications, and the possible repercussions of not taking them as prescribed to avoid these interactions.
Individual patient characteristics, such as age, health status, food preferences, and genetics, should be taken into account when creating treatment plans. There might be more practical options for medications that have fewer interactions with food. It is important to regularly check blood drug levels and nutritional status to spot and treat any possible nutrient imbalances or deficiencies [81]. Using genetic data to manage drug-food interactions can assist in customizing interventions based on the genetic profile of each patient.
The three methods of drug absorption are facilitated diffusion, active transport, and passive diffusion. Drug characteristics affect passive diffusion, but carrier proteins are needed for active transport to promote absorption. The “food effect” of high-fat meals, particularly grapefruit juice, can alter how well drugs are absorbed. Certain nutrients or food ingredients, like grapefruit juice, can inhibit or induce cytochrome P450 enzymes in the liver, which can change the concentration of drugs in the bloodstream and have a substantial impact on drug metabolism. This may lead to modifications in drug clearance and possible adjustments in therapeutic outcomes.
Increased solubility and absorption, leading to higher drug concentrations, can result in increased efficacy, whereas inappropriate drug absorption during meals can compromise drug absorption. Elevated drug concentrations can increase the risk of side effects and toxicity due to changes in drug absorption and metabolism.
Certain drug-food interactions may require dietary restrictions, which could affect patient compliance. Individual reactions to drug-food interactions can also be influenced by genetic variability, and age-related alterations in gastrointestinal physiology can affect how well drugs are absorbed (Figure 10) [82].

Figure 10: Healthcare providers must consider drug-food interactions, genetic factors, and dietary restrictions to optimize prescription regimens and maintain healthy diets, requiring open communication and multidisciplinary advice.
Discussion
Monitoring and management of interactions
This guide emphasizes the importance of monitoring and managing drug interactions in healthcare settings to ensure safe and efficient medication use. Regular monitoring can prevent negative outcomes and maintain therapeutic goals, while promoting a collaborative approach to treatment [83].
Drug interactions can be identified through a thorough medication review, pharmacogenomic testing, patient monitoring, and cooperation between healthcare providers. This involves examining a patient’s entire medication history, including supplements. Personalized medication management can be facilitated by pharmacogenomic testing, which detects genetic variations. Regular monitoring and reporting of symptoms and side effects can help identify potential interactions early [84].
Patient education is important for effective medication management, emphasizing the importance of a thorough medical history and following recommended dosage schedules. Collaborative decision- making promotes communication among medical professionals and involves patients in treatment discussions. However, managing medication interactions in polypharmacy, characterized by multiple chronic illnesses, can be challenging due to complicated regimens, adherence issues, and limited data. Barriers like language barriers, health literacy, and limited patient understanding also hinder prompt identification. Despite these challenges, improvements in pharmacogenomics can lead to more modified drug regimens and improved drug interaction prediction [85]. Remote monitoring and telehealth technologies can enable continuous monitoring, allowing medical professionals to recognize and manage interactions from a distance. Mobile health apps and on-demand medical assistance can help patients actively monitor and manage their medications.
The Health Information Exchange (HIE) can enhance healthcare interoperability by facilitating comprehensive medication reviews and teamwork. Data analytics can inform healthcare policies, while professional development programs can enhance knowledge about drug interactions. Public health campaigns can raise awareness about the importance of informing healthcare providers about medications and supplements [86].
Continuous monitoring for potential interactions: Continuous monitoring is important in modern healthcare to optimize therapeutic results, prevent adverse events, and ensure patient safety. It considers the fluid nature of patient care, adjusting prescriptions, health, and lifestyle choices. It helps manage polypharmacy, managing drug regimens for patients with multiple chronic illnesses. It allows for drug schedule changes due to new diseases or recovery [87]. Continuous monitoring ensures medical professionals are aware of new risks and aligns with patient-centered care, focusing on personalized medication administration.
Continuous monitoring of drug schedules can be achieved through various techniques and instruments, including periodic reviews, patient interviews, Electronic Health Records (EHRs), PIMs, Clinical Decision Support Systems (CDSS), medication therapy management, pharmacy information systems, pharmacogenomic testing, adverse event reporting systems, and genetic screening [88]. Real-time monitoring of prescription orders by pharmacy information systems allows pharmacists to collaborate with prescribers and intervene when possible interactions are identified. CDSS offers evidence-based suggestions for different drugs or dose modifications, while pharmacogenomic testing identifies genetic variations influencing drug metabolism. However, healthcare continuous monitoring faces challenges such as patient adherence, polypharmacy complexity, resource limitations, incomplete data, interaction variability, and reluctance towards technology. Future directions in continuous monitoring include the integration of cutting-edge technologies like AI, blockchain, wearable technology, mobile applications, patient-generated health data, population health analytics, and predictive analytics [89].
Healthcare providers can enhance the efficiency and efficacy of continuous monitoring practices by standardizing data formats, creating national health information exchanges, improving health literacy, promoting patient-provider shared decision-making, and utilizing telehealth platforms. These initiatives can improve interoperability, patient involvement, and remote access to medical staff, ultimately leading to more effective and efficient healthcare services (Figure 11).

Figure 11: (A) Continuous monitoring in healthcare optimizes therapeutic results, prevents adverse events, and ensures patient safety; (B) Techniques include periodic reviews, patient interviews, EHRs, CDSS, and genetic screening; (C) Challenges include patient adherence, polypharmacy complexity, resource limitations, and data inconsistencies.
Adjusting dosages or changing medications to minimize adverse effects: Adverse effects in pharmacotherapy can hinder treatment outcomes and patient safety. Individual reactions to medications can vary, necessitating customized dosing plans. Suboptimal response can affect therapy efficacy, necessitating dosage modifications guided by therapeutic drug monitoring [90]. Health status changes can affect drug metabolism, necessitating dosage adjustments. Medication interactions can also affect efficacy and metabolism.
Healthcare practitioners play a vital role in directing these choices and ensuring patients are informed about the importance of medication. Principles such as patient-centered care, Therapeutic Drug Monitoring (TDM), baseline health status, individualized patient assessment, and routine monitoring and follow-up are used to adjust dosages in the healthcare industry. Decisions are based on a thorough medical history, including allergies, past adverse reactions, and medication history [91]. TDM is particularly helpful for medications with narrow therapeutic indices, where minute dosage adjustments can significantly impact outcomes. Patient- centered care encourages shared decision-making and patient education, while frequent monitoring and follow-up visits help evaluate the effectiveness of dosage adjustments. Pharmacokinetics determine the best dosage schedules, and healthcare professionals work together to optimize therapy, minimize psychological impact, and provide emotional support for patients enduring negative effects.
Healthcare professionals guide patients through this process, improving safety and effectiveness with precision medicine and digital health solutions [92]. Refinement and optimization through research, education, and technology enhance patient outcomes and quality of life.
Conclusion
Drug development is a complex and time-consuming process that takes 14 years to complete. Advancements in combinatorial chemistry and high-throughput screening methods have accelerated the process, with Computer-Aided Drug Design (CADD) being a popular approach. The process involves seven steps: disease selection, target selection, lead compound identification, lead optimization, preclinical trial testing, clinical trial testing, and pharmacogenomic optimization. Protein function modulators require biological targets to be druggable, safe, and meet clinical requirements.
Clinical trials are research investigations where volunteers help find solutions to specific health issues, overseen by the FDA. Successful NDA approval can lead to market access, clinical impact, investment returns, and scientific advancement. Challenges include technological advancements, regulatory compliance, quality assurance, and data integration. Future trends include machine learning, artificial intelligence, real-time data monitoring, and patient-centric strategies.
Regulatory approval is the formal authorization to market, sell, or use a new healthcare product, ensuring it meets established standards for quality, safety, and efficacy. Regulatory bodies like TGA, PMDA, EMA, and FDA monitor approvals. Post-Marketing Surveillance (PMS) is important for managing medications, medical equipment, and healthcare products, ensuring product safety, evaluating adverse events, and guiding regulatory decisions. Challenges include underreporting, bias, data quality, and global variability.
Future developments include incorporating empirical data, patient- centered strategies, cutting-edge technology, and international cooperation. Continuous monitoring is essential for identifying hidden risks, evaluating effectiveness, and validating clinical trials. Understanding drug interactions is important for determining a drug’s concentration and predicting its physiological effects.
Pharmacodynamics is a field of pharmacology that studies how medications affect the body and their interactions with receptors, enzymes, or ion channels. It focuses on the therapeutic index, which measures a medication’s safety margin. Genetics, environment, and medical conditions also influence medication reactions. Personalized medicine uses pharmacogenomics to study genetic differences affecting medication response. Synergistic effects between drugs can result from receptor activation, complementary activities, or pharmacokinetic interactions. Antagonism occurs when two or more drugs interact, resulting in reduced effects. Drug- drug interactions can lead to increased toxicity or decreased efficacy. Polypharmacy involves concurrent use of multiple medications to increase efficacy or reduce adverse effects. Open communication and a multidisciplinary team are essential for personalized care. Continuous monitoring, therapeutic drug monitoring, and patient- centered care are essential for adjusting dosages and minimizing side effects.
Acknowledgments
The authors would like to thank Mr. Vikas Srivastava, Assistant professor, Department of Applied Sciences (RRIMT) for providing the necessary information technology related resources to carry out the project.
Conflicts of Interest
There is no Conflict of Interest among Authors.
References
- Amarji B, Abed SN, Bairagi U, Deb PK, Al-Attraqchi O, Choudhury AA, et al. Four stages of pharmaceutical product development: Preformulation, prototype development and scale-up, biological aspects, and commercialization. In Dosage Form Design Considerations. 2018:637-668.
- Gouda AM, Ali HI, Almalki WH, Azim MA, Abourehab MA, Abdelazeem AH. Design, synthesis, and biological evaluation of some novel pyrrolizine derivatives as COX inhibitors with anti-inflammatory/analgesic activities and low ulcerogenic liability. Molecules. 2016;21(2):201.
[Crossref] [Google Scholar] [PubMed]
- Prieto-Martínez FD, López-López E, Juárez-Mercado KE, Medina-Franco JL. Computational drug design methods—current and future perspectives. In Silico Drug Design. 2019:19-44.
- Guruprasad L, Andola P, Banerjee A, Laxman D, Naresh GK. Structure-based methods in drug design. In Cheminformatics, QSAR and Machine Learning Applications for Novel Drug Development 2023:205-237.
- Wang S, Sim TB, Kim YS, Chang YT. Tools for target identification and validation. Curr Opin Chem Biol. 2004;8(4):371-377.
[Crossref] [Google Scholar] [PubMed]
- Caldwell GW. In silico tools used for compound selection during target-based drug discovery and development. Expert Opin Drug Discov. 2015;10(8):901-923.
[Crossref] [Google Scholar] [PubMed]
- Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M, et al. Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem. 2008;51(3):347-372.
[Crossref] [Google Scholar] [PubMed]
- Roy A. High-Throughput Screening (HTS) Technology. In Encyclopaedia of Molecular Pharmacology. 2022:787-799.
- Erlanson DA, Jahnke W. Fragment-based approaches in drug discovery. Wiley-VCH. 2006.
- Tammela P. Screening Methods for the Evaluation of Biological Activity in Drug Discovery. 2004.
- Di L, Kerns EH. Drug-like properties: Concepts, structure design and methods from ADME to toxicity optimization. Academic press. 2015.
- Mignani S, Huber S, Tomas H, Rodrigues J, Majoral JP. Compound high-quality criteria: A new vision to guide the development of drugs, current situation. Drug Discov Today. 2016;21(4):573-584.
[Crossref] [Google Scholar] [PubMed]
- Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502(7472):480-488.
[Crossref] [Google Scholar] [PubMed]
- DeWitt DS, Hawkins BE, Dixon CE, Kochanek PM, Armstead W, Bass CR, et al. Pre-clinical testing of therapies for traumatic brain injury. J Neurotrauma. 2018;35(23):2737-2754.
[Crossref] [Google Scholar] [PubMed]
- Lock RB, Liem NL, Papa RA. Preclinical testing of antileukemic drugs using an in vivo model of systemic disease. Methods Mol Med. 2005;111:323-334.
[Crossref] [Google Scholar] [PubMed]
- Caponigro G, Sellers WR. Advances in the preclinical testing of cancer therapeutic hypotheses. Nat Rev Drug Discov. 2011;10(3):179-187.
[Crossref] [Google Scholar] [PubMed]
- An F, Qu Y, Luo Y, Fang N, Liu Y, Gao Z, et al. A laminated microfluidic device for comprehensive preclinical testing in the drug ADME process. Sci Rep. 2016;6(1):25022.
[Crossref] [Google Scholar] [PubMed]
- Holbein MB. Understanding FDA regulatory requirements for investigational new drug applications for sponsor-investigators. J Investig Med. 2009;57(6):688-694.
[Crossref] [Google Scholar] [PubMed]
- Gawai AA, Shaikh F, Gadekar M, Deokar N, Kolhe S, Biyani KR. A review on: Phase ‘0’clinical trials or exploratory investigational new drug. Turk J Pharm Sci. 2017;14(1):84-89.
[Crossref] [Google Scholar] [PubMed]
- Nugent P, Duncan JN, Colagiovanni DB. Preparation of a preclinical dossier to support an Investigational New Drug (IND) application and first-in-human clinical trial. In A Comprehensive Guide to Toxicology in Nonclinical Drug Development. 2017:189-213.
- Kummar S, Rubinstein L, Kinders R, Parchment RE, Gutierrez ME, Murgo AJ, et al. Phase 0 clinical trials: Conceptions and misconceptions. Cancer J. 2008;14(3):133-137.
[Crossref] [Google Scholar] [PubMed]
- Simon R, Wittes RE, Ellenberg SS. Randomized phase II clinical trials. Cancer Treat Rep. 1985;69(12):1375-1381.
[Google Scholar] [PubMed]
- Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3(1):24-40.
[Crossref] [Google Scholar] [PubMed]
- Freidlin B, Sun Z, Gray R, Korn EL. Phase III clinical trials that integrate treatment and biomarker evaluation. J Clin Oncol. 2013;31(25):3158.
[Crossref] [Google Scholar] [PubMed]
- Monahan C, Babiarz JC. The new drug application. In FDA Regulatory Affairs. 2008:79-118.
- Shah DN. Obtaining approval of new drug applications and abbreviated new drug applications from a chemistry, manufacturing, and controls perspective. In The Pharmaceutical Regulatory Process. 2004:417-438.
- DiMasi JA. A new look at United States drug development and approval times. Am J Ther. 1996;3(9):647-557.
[Crossref] [Google Scholar] [PubMed]
- Eifler AC, Thaxton CS. Nanoparticle therapeutics: FDA approval, clinical trials, regulatory pathways, and case study. Methods Mol Biol. 2011:325-338.
[Crossref] [Google Scholar] [PubMed]
- Wang J, Chow SC. On the regulatory approval pathway of biosimilar products. Pharmaceuticals (Basel). 2012;5(4):353-368.
[Crossref] [Google Scholar] [PubMed]
- Wilks SJ, Hara SA, Ross EK, Nicolai EN, Pignato PA, Cates AW, et al. Non-clinical and pre-clinical testing to demonstrate safety of the barostim neo electrode for activation of carotid baroreceptors in chronic human implants. Front Neurosci. 2017;11:274579.
[Crossref] [Google Scholar] [PubMed]
- Hansson MG. Building on relationships of trust in biobank research. J Med Ethics. 2005;31(7):415-418.
[Crossref] [Google Scholar] [PubMed]
- Marcus HJ, Payne CJ, Hughes-Hallett A, Marcus AP, Yang GZ, Darzi A, et al. Regulatory approval of new medical devices: Cross sectional study. BMJ. 2016;353.
[Crossref] [Google Scholar] [PubMed]
- Kaplan AV, Baim DS, Smith JJ, Feigal DA, Simons M, Jefferys D, et al. Medical device development: From prototype to regulatory approval. Circulation. 2004;109(25):3068-3072.
[Crossref] [Google Scholar] [PubMed]
- Wang J, Chow SC. On the regulatory approval pathway of biosimilar products. Pharmaceuticals (Basel). 2012;5(4):353-368.
[Crossref] [Google Scholar] [PubMed]
- Greene SM, Geiger AM. A review finds that multicentre studies face substantial challenges but strategies exist to achieve Institutional Review Board approval. J Clin Epidemiol. 2006;59(8):784-790.
[Crossref] [Google Scholar] [PubMed]
- Amdur RJ, Biddle C. Institutional review board approval and publication of human research results. JAMA. 1997;277(11):909-914.
[Crossref] [Google Scholar] [PubMed]
- Dabrowska A, Thaul S. How FDA approves drugs and regulates their safety and effectiveness. Washington: Congressional Research Service. 2018:1-25.
- Ciociola AA, Cohen LB, Kulkarni P, Kefalas C, Buchman A, Burke C, et al. How drugs are developed and approved by the FDA: Current process and future directions. Am J Gastroenterol. 2014;109(5):620-623.
[Crossref] [Google Scholar] [PubMed]
- Lipsky MS, Sharp LK. From idea to market: The drug approval process. J Am Board Fam Pract. 2001;14(5):362-367.
[Google Scholar] [PubMed]
- Guarino RA. New drug approval process. Drugs and the pharmaceutical sciences. 1993;190:190.
- Carmo AC, Piras SS, Rocha NF, Gratieri T. Main reasons for registration application refusal of generic and similar pharmaceutical drug products by the Brazilian Health Regulatory Agency (ANVISA). Biomed Res Int.2017.
[Crossref] [Google Scholar] [PubMed]
- Junod V. Drug marketing exclusivity under United States and European Union law. Food Drug Law J. 2004;59(4):479-518.
[Google Scholar] [PubMed]
- Gough S. Post-marketing surveillance: A UK/European perspective. Curr Med Res Opin. 2005;21(4):565-570.
[Crossref] [Google Scholar] [PubMed]
- Westerholm B. The rationale for a post-marketing surveillance. Hum Reprod. 1987;2(1):41-44.
[Crossref] [Google Scholar] [PubMed]
- Jones DC, Langman MJ, Lawson DH, Vessey MP. Post-marketing surveillance of the safety of cimetidine: Twelve-month morbidity report. Q J Med. 1985;54(3-4):253-268.
[Crossref] [Google Scholar] [PubMed]
- Cohen S, Curtis JR, DeMasi R, Chen Y, Fan H, Soonasra A, et al. Worldwide, 3-year, post-marketing surveillance experience with tofacitinib in rheumatoid arthritis. Rheumatol Ther. 2018;5:283-291.
[Crossref] [Google Scholar] [PubMed]
- Beninger P. Pharmacovigilance: An overview. Clin Ther. 2018;40(12):1991-2004.
[Crossref] [Google Scholar] [PubMed]
- Löscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: Ways out of the current dilemma. Epilepsia. 2011;52(4):657-678.
[Crossref] [Google Scholar] [PubMed]
- Howard DH, Bach PB, Berndt ER, Conti RM. Pricing in the market for anticancer drugs. J Econ Perspect. 2015;29(1):139-162.
[Crossref] [Google Scholar] [PubMed]
- Van Zee A. The promotion and marketing of oxycontin: Commercial triumph, public health tragedy. Am J Public Health. 2009;99(2):221-227.
[Crossref] [Google Scholar] [PubMed]
- Palleria C, Di Paolo A, Giofrè C, Caglioti C, Leuzzi G, Siniscalchi A, et al. Pharmacokinetic drug-drug interaction and their implication in clinical management. J Res Med Sci. 2013;18(7):601.
[Google Scholar] [PubMed]
- Prichard MN, Shipman Jr C. A three-dimensional model to analyse drug-drug interactions. Antiviral Res. 1990;14(4-5):181-205.
[Crossref] [Google Scholar] [PubMed]
- Bahramsoltani R, Rahimi R, Farzaei MH. Pharmacokinetic interactions of curcuminoids with conventional drugs: A review. J Ethnopharmacol. 2017;209:1-12.
[Crossref] [Google Scholar] [PubMed]
- Benet LZ, Kroetz D, Sheiner L, Hardman J, Limbird L. Pharmacokinetics: The dynamics of drug absorption, distribution, metabolism, and elimination. Goodman and Gilman’s the pharmacological basis of therapeutics. 1996;3:e27.
- Lin J, Sahakian DC, De Morais SM, Xu JJ, Polzer RJ, Winter SM. The role of absorption, distribution, metabolism, excretion and toxicity in drug discovery. Curr Top Med Chem. 2003;3(10):1125-1154.
[Crossref] [Google Scholar] [PubMed]
- Kok-Yong S, Lawrence L, Ahmed T. Drug distribution and drug elimination. In Basic pharmacokinetic concepts and some clinical applications. 2015:99-116.
- Shanu-Wilson J, Evans L, Wrigley S, Steele J, Atherton J, Boer J. Biotransformation: Impact and application of metabolism in drug discovery. ACS Med Chem Lett. 2020;11(11):2087-2107.
[Crossref] [Google Scholar] [PubMed]
- Wang L, Huang Q, Wu J, Wu W, Jiang J, Yan H, et al. The metabolism and biotransformation of AFB1: Key enzymes and pathways. Biochem Pharmacol. 2022;199:115005.
[Crossref] [Google Scholar] [PubMed]
- Jangoux M. Excretion. In Echinoderm nutrition. 2020:437-445.
- Rollins DE, Klaassen CD. Biliary excretion of drugs in man. Clin Pharmacokinet. 1979;4:368-379.
[Crossref] [Google Scholar] [PubMed]
- Taft DR. Drug excretion. In Pharmacology. 2009:175-199.
- Jaap Vuyk MD. Pharmacokinetic and pharmacodynamic interactions between opioids and propofol. J Clin Anesth. 1997;9(6):23S-6S.
[Crossref] [Google Scholar] [PubMed]
- Verrotti A, Lattanzi S, Brigo F, Zaccara G. Pharmacodynamic interactions of antiepileptic drugs: From bench to clinical practice. Epilepsy Behav. 2020;104:106939.
[Crossref] [Google Scholar] [PubMed]
- Niu J, Straubinger RM, Mager DE. Pharmacodynamic drug–drug interactions. Clin Pharmacol Ther. 2019;105(6):1395-406.
[Crossref] [Google Scholar] [PubMed]
- Duff C. The place and time of drugs. Int J Drug Policy. 2014;25(3):633-639.
[Crossref] [Google Scholar] [PubMed]
- Michael M, Rosengarten M. Medicine: Experimentation, politics, emergent bodies. Body Soc. 2012;18(3-4):1-17.
- Hogle LF. Recovering the nation's body: Cultural memory, medicine, and the politics of redemption. Rutgers University Press. 1999.
- Ho YS, So KF, Chang RC. Anti-aging herbal medicine—how and why can they be used in aging-associated neurodegenerative diseases?. Ageing Res Rev. 2010;9(3):354-562.
[Crossref] [Google Scholar] [PubMed]
- Yin N, Ma W, Pei J, Ouyang Q, Tang C, Lai L. Synergistic and antagonistic drug combinations depend on network topology. PloS One. 2014;9(4):e93960.
[Crossref] [Google Scholar] [PubMed]
- Tekin E, Beppler C, White C, Mao Z, Savage VM, Yeh PJ. Enhanced identification of synergistic and antagonistic emergent interactions among three or more drugs. J R Soc Interface. 2016;13(119):20160332.
[Crossref] [Google Scholar] [PubMed]
- Tome M, Zupan J, Tomičić Z, Matos T, Raspor P. Synergistic and antagonistic effects of immunomodulatory drugs on the action of antifungals against Candida glabrata and Saccharomyces cerevisiae. PeerJ. 2018;6:e4999.
[Crossref] [Google Scholar] [PubMed]
- Prichard MN, Shipman Jr C. A three-dimensional model to analyse drug-drug interactions. Antiviral Res. 1990;14(4-5):181-205.
[Crossref] [Google Scholar] [PubMed]
- Björkman IK, Fastbom J, Schmidt IK, Bernsten CB, Pharmaceutical Care of the Elderly in Europe Research (PEER) Group. Drug—drug interactions in the elderly. Ann Pharmacother. 2002;36(11):1675-1681.
[Crossref] [Google Scholar] [PubMed]
- Percha B, Altman RB. Informatics confronts drug–drug interactions. Trends Pharmacol Sci. 2013;34(3):178-184.
[Crossref] [Google Scholar] [PubMed]
- Bijnsdorp IV, Giovannetti E, Peters GJ. Analysis of drug interactions. Methods Mol Biol. 2011:421-434.
[Crossref] [Google Scholar] [PubMed]
- Pennings EJ, Leccese AP, Wolff FA. Effects of concurrent use of alcohol and cocaine. Addiction. 2002;97(7):773-783.
[Crossref] [Google Scholar] [PubMed]
- Pandey A, Mishra AK. Immunomodulation, toxicity, and therapeutic potential of nanoparticles. BioTech (Basel). 2022;11(3):42.
[Crossref] [Google Scholar] [PubMed]
- Brand W, Noorlander CW, Giannakou C, De Jong WH, Kooi MW, Park MV, et al. Nanomedicinal products: A survey on specific toxicity and side effects. Int J Nanomedicine. 2017:6107-6129.
[Crossref] [Google Scholar] [PubMed]
- Yamreudeewong W, Henann NE, Fazio A, Lower DL, Cassidy TG. Drug-food interactions in clinical practice. J Fam Pract. 1995;40(4):376-385.
[Google Scholar] [PubMed]
- Schmidt LE, Dalhoff K. Food-drug interactions. Drugs. 2002;62:1481-1502.
[Crossref] [Google Scholar] [PubMed]
- Otles S, Senturk A. Food and drug interactions: A general review. Acta Sci Pol Technol Aliment. 2014;13(1):89-102.
[Google Scholar] [PubMed]
- Rajan R, Roy AD, Todi S, Dutta P, Chatterjee T, Lahiri S, et al. A review article on impact of drug food interactions and its prevention. Int J Food Sci Nutr. 2018;3(4).
- Foy M, Sperati CJ, Lucas GM, Estrella MM. Drug interactions and antiretroviral drug monitoring. Curr HIV/AIDS Rep. 2014;11:212-222.
[Crossref] [Google Scholar] [PubMed]
- Gex-Fabry M, Balant LP, Balant-Gorgia AE. Therapeutic drug monitoring databases for post-marketing surveillance of drug-drug interactions. Drug Saf. 2001;24:947-959.
[Crossref] [Google Scholar] [PubMed]
- Kang JS, Lee MH. Overview of therapeutic drug monitoring. Korean J Intern Med. 2009;24(1):1.
[Crossref] [Google Scholar] [PubMed]
- Munnink TH, Demmer A, Slenter RH, Movig KL. Amiodarone rifampicin drug–drug interaction management with therapeutic drug monitoring. Ther Drug Monit. 2018;40(2):159-161.
[Crossref] [Google Scholar] [PubMed]
- Bourgeois W, Hogben P, Pike A, Stuetz RM. Development of a sensor array based measurement system for continuous monitoring of water and wastewater. Sens Actuators B Chem. 2003;88(3):312-319.
- Della Seta M, Esposito C, Fiorucci M, Marmoni GM, Martino S, Sottili G, et al. Thermal monitoring to infer possible interactions between shallow hydrothermal system and slope-scale gravitational deformation of Mt Epomeo (Ischia Island, Italy). Geol Soc Spec Publ. 2024;519(1):5-27.
- Järvi J, Hannuniemi H, Hussein T, Junninen H, Aalto PP, Hillamo R, et al. The urban measurement station SMEAR III: Continuous monitoring of air pollution and surface–atmosphere interactions in Helsinki, Finland. Boreal Environ Res. 2009;14:86.
- Turnheim K. When drug therapy gets old: Pharmacokinetics and pharmacodynamics in the elderly. Exp Gerontol. 2003;38(8):843-853.
[Crossref] [Google Scholar] [PubMed]
- Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: A randomized controlled trial. Neurology. 2006;66(3):366-372.
[Crossref] [Google Scholar] [PubMed]
- Hanahan D, Bergers G, Bergsland E. Less is more, regularly: Metronomic dosing of cytotoxic drugs can target tumor angiogenesis in mice. J Clin Invest. 2000;105(8):1045-1047.
[Crossref] [Google Scholar] [PubMed]
Citation: Maurya N, Verma A, Kumar D, Ayushi (2024) A Critical Examination of Drug Discovery and its Complex Interplay with the Human Body: A Review. J Clin Exp Pharmacol. 14.401.
Copyright: © 2024 Maurya N, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.