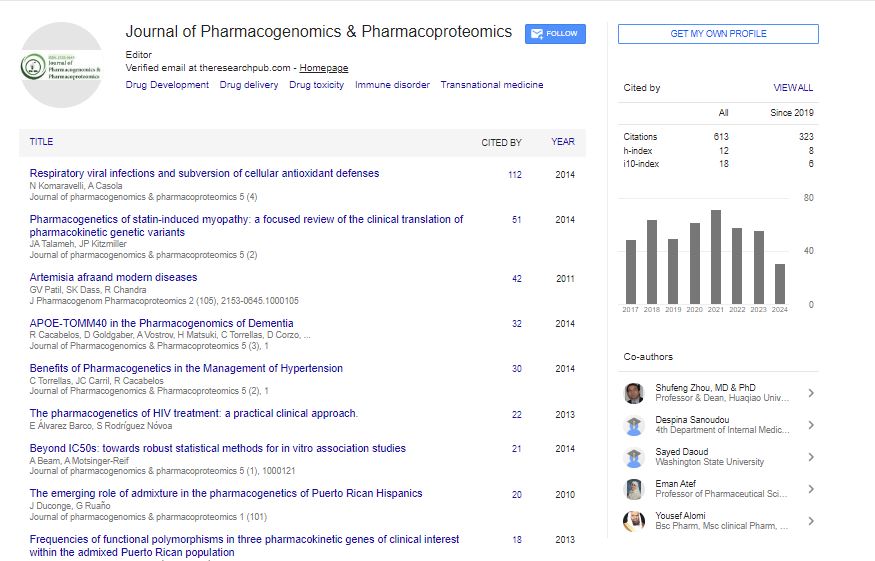

Indexed In
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- JournalTOCs
- ResearchBible
- Electronic Journals Library
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Proquest Summons
- SWB online catalog
- Virtual Library of Biology (vifabio)
- Publons
- MIAR
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Review Article - (2023) Volume 14, Issue 1
Pharmacogenetics of Second Generation Antihistamines: A Systematic Review
Asees Singh*Received: 13-Jul-2022, Manuscript No. JPP-22-17433; Editor assigned: 15-Jul-2022, Pre QC No. JPP-22-17433 (PQ); Reviewed: 29-Jul-2022, QC No. JPP-22-17433; Revised: 23-Dec-2022, Manuscript No. JPP-22-17433 (R); Published: 04-Jan-2023, DOI: 10.35248/2153-0645.22.14.31
Abstract
H1 inverse agonists, more commonly known as anti-histamines, find great everyday use in our lives. They function by blocking the H1 receptor and not allowing histamine to express its physiological function. They are mostly used for the treatment of allergies, rhinitis etc. The second generation of this class of drugs is different from the first because they exist in a zwitterionic form at the physiological pH, thus not crossing the Blood Brain Barrier (BBB) and not showing CNS symptoms, like drowsiness. This article reviews how the reported genetic variations in the population alter the pharmacology of these drugs. With rising cases of minor allergies, especially reported in polluted cities like Delhi, a pharmacogenetic approach to prescribing anti-histamines can lead to better and more targeted treatment of the symptoms. The paper covers both the pharmacokinetic parameters and the pharmacodynamic parameters and then goes on to discuss how genetic variations have been reported in the genes encoding for the proteins affecting said parameters.
Keywords
Pharmacogenomics; Antihistamines; Second Generation Antihistamines (SGAs); Blood Brain Barrier (BBB)
INTRODUCTION
Antihistamines, technically referred to as H1 antagonists or more precisely H1 inverse agonists competitively antagonize actions of histamine at the H1 receptors. Recent evidence indicates that the histamine H1 receptor exhibits some degree of constitutive activity at certain sites and few H1 antagonists are also inverse agonists. The first H1 antagonists were introduced in the late 1930’s and have subsequently proliferated into unnecessary motley of drugs. Nevertheless, they are frequently used for a variety of purposes. More commonly employed now are the less sedating/no sedating second generation H1 antihistamines added after 1980. Seemingly, H1 antihistaminic have diverse chemical structures, but the majority has a substituted ethylamine side chain [1].
Pharmacologically, a drug undergoes two aspects; what the body does to the drug, i.e., pharmacokinetics and what the drug does to the body, i.e., pharmacodynamics. Our genetic makeup can alter both the kinetics and the dynamics of a drug, depending on how impactful the variation is and how much of the variant gene is expressed in the body. This impact and expression levels are moderated by the location of the gene, the regulators, any linkage with another gene and various epigenetic factors. The study of these variations and their impact on pharmacology is what pharmacogenetics.
Literature Review
We will be considering the common Second Generation Antihistamines (SGAs) and will be reviewing the variations reported that can affect each pharmacologically relevant process and parameter of the drug. The SGAs in review will be:
• Acrivastine
• Astemizole
• Bepotastine
• Bilastine
• Cetirizine
• Fexofenadine
• Ketotifen
• Levocetirizine
• Loratidine
• Mizolastine
• Rupatadine
The other SGAs, like Ebastine, Terfenadine etc. have either been withdrawn due to side effects like long QT syndrome or are prescribed in a very specific demographic, where variation in the gene pool would be minimal and has thus not been studied.
Pharmacogenetic review
Pharmacokinetics: Four main processes define the pharmacokinetics of a drug; absorption, distribution, metabolism and excretion
Absorption and distribution: When we consider genetic variations reported concerning the pharmacokinetics of SGAs, we find that absorption has mostly remained unchanged and has mostly been a function of the formulation used and the route of administration. The bioavailability has not been markedly altered or relevant trials for ascertaining a marked change have not been done. Thus, we can safely assume that there is no real impact of genetics on the absorption process.
Distribution is a function of the plasma protein binding efficiency of the drug. The effect of SNPs and polymorphisms in the human plasma protein genes has been studied in 2013 by Johansson et al. [2]. Their discoveries, however, have not been correlated to the plasma binding of SGAs. Thus, their protein binding has not been studied as a function of the variations in the human plasma proteome.
Metabolism: Considering the metabolism of the SGAs in question, the following table can be tabulated, containing the enzymes involved in each SGA’s metabolism (Table 1).
| Drug | Metabolizing enzymes |
|---|---|
| Astemizole | CYP3A4, CYP2D6 |
| Bepotastine | CYP3A4, CYP2C9, CYP2C19 |
| Ketotifen | UGT1A3, UGT1A4, UGT2B10 |
| Levocetirizine | Amine Oxidases, UGT enzymes, cholyl-CoA synthetase, GSH-S-Transferases |
| Mizolastine | Sulfotransferases |
| Rupatadine | CYP3A4, CYP2C9, CYP2C19, CYP2D6 |
Abbreviations: CYPxxx: P450 Cytochrome xxx; UGTxxx: UDP-glucosyltransferase xxx; CoA:Coenzyme A; GSH: Glutathione
Table 1: Enzymes involved in the metabolism of SGAs.
The other drugs discussed in the introduction, like Fexofenadine, Cetirizine and Bilastine are not metabolized or the metabolized percentage is less than equal to 10%. The metabolism of Acrivastine has not been studied [3-10].
P450 Cytochrome family (CYPxxx)
The P450 cytochrome family is the major metabolizing family involved in the metabolism of all xenobiotics. Denoted by the CYP, (Cytochrome P450), this family is a hemoprotein based oxidizing enzyme group, which is named with the help of the following abbreviation method; CYP+Arabic Numeral+Letter +Arabic Numeral. All the isoforms are significant in our body’s metabolism, but we will focus our discussion on the isoforms involved in the metabolism of SGAs.
CYP3A4
CYP3A4 is the most versatile and influential enzyme involved in the metabolism of substrates present inside our body and those administered to it. The variation in CYP3A4’s activity is so dynamic and so non-specific to one cause, that we have not been able to place a finger on it. A lot of environmental, genetic and epigenetic factors have been identified. Focussing on the genetic factors, the SNPs discovered in the CYP3A4 locus located on chromosome 7 have not been able to explain phenotypic variability to great extent [11].
In the year 2000, Ozdemir et al. showed that the genetic control over the action of CYP3A4 was determined by the substrate in question. It was even found that the time of day affected the control the genetic makeup had over CYP3A4’s actions [12].
With the advent of the ‘1000 genome project’, there is hope concerning the effect of SNPs inside the CYP3A4 locus. The SNPs were able to explain plasma concentrations of a different drug when rare alleles were together with low expressor *6 allele [13].
In summary, we can say that the modulation of CYP3A4 by the genetic makeup of the CYP3A4 gene and the associated regulatory genes is only part of the picture in terms of the variable behaviour of CYP3A4. With the research in the ‘1000 genome project’ and experimental interpretation of the collected data, we would have a better understanding of the genetic component of the regulation of CYP3A4 activity [14].
CYP2C9
CYP2C9 is another important member of the P450 cytochrome superfamily of enzymes. The gene for coding the CYP2C9 enzyme is located on the long arm of the 10th chromosome. Approximately 60 allelic variants of the CYP2C9 gene have been reported, with the CYP2C9*2 and CYP2C9*3 being the most studied alleles. Studies on S-warfarin have shown that CYP2C9*2 and CYP2C9*3 inhibit catalytic activity. Other alleles which inhibit metabolism and have been studied are CYP2C9*5, CYP2C9*6, CYP2C9*8 and CYP2C9*11. The distribution of these alleles varies demographically across the world, with CYP2C9*2 and CYP2C9*3 being found in Caucasians, CYP2C9*6, CYP2C9*8 being found in African- Americans [15].
CYP2C19
CYP2C19 metabolizes many drugs ingested by us. The encoding gene is located on the long arm of chromosome 10, in a locus of P450 cytochrome enzymes.
Of the variants studied, CYP2C19*2 and CYP2C19*3 are defective, due to a splice defective site and a premature stop codon respectively. The variants have been classified as Poor Metabolizers (PMs). The newly discovered CYP2C19*17, upon various studies, was discovered to be marginally better than the extensive metabolizing class of variants of CYP2C19, as reviewed by Wan-Po [16].
As a result, we can say that there are certain alleles of CYP2C19 which have been characterized as Ultrarapid Metabolizers (UM), some as extensive and some as poor, like CYP2C19*2 and CYP2C19*3
CYP2D6
Being one of the most investigated CYP family enzymes, we have discovered approximately 74 allelic variants of the CYP2D6 enzyme. They have been classified as PMs, UMs and Extensive Metabolizers (EMs).
The given table shows the distribution of the discussed variants among the various types of metabolizers (Table 2).
| Extensive metabolizers | Ultra metabolizers | Poor metabolizers |
|---|---|---|
| *1 | - | No activity |
| *3, *4, *5, *6, *7, *8, *11, *12, *13, *14, *15, *16, *18, *19, *20, *21, *38, *40, *42, *44, *56, *62 | ||
| Substrate depended activity | ||
| *10, *17, *36, *41 |
Table 2: Classification of all allelic variants of CYP2D6 on the basis of metabolizing activity.
Apart from those mentioned in the table, CYP2D6*2 and CYP2D6*36 are important [17].
UDP-Glucuronosyltransferases (UGTxxx)
UGT enzymes are those which are involved in the main phase 2 metabolism reaction, glucuronic acid conjugation. Glucuronic acid is the preferred conjugate for many drugs in the body due to its high supply from the Krebs cycle and its ease of attachment.
As of 2002, genetic polymorphism in the UGT superfamily has been reported for 6 of the 16 enzymes, namely UGT1A1, UGT1A6, UG1A7, UGT2B4, UGT2B7 and UGT2B15. Functional inhibition has only been reported for UGT1A1 [18].
UGT1A3
In 2004, Iwai et al. conducted studies on the Japanese population and found out that UGT1A3 exists as 6 different allelic variants in the population [19]. Of these, 4 caused amino acid substitution and 2 were silent. The variation resulted in changed enzyme efficiency, with the W11R showing 121% activity of the wild type and the W11R-V47A variant showing 369%. The other 4 showed reduced activity.
UGT1A4
Over 100 polymorphisms have been reported in the UGT1A4 gene, until 2014. Of note are the two variants UGT1A4*2, associated with decreased activity and UGT1A4*3, associated with increased activity. UGT1A4*3 has leucine to valine at codon 48, whereas UGT1A4*2 has threonine from proline [20].
UGT2B10
As per the Ensembl database, one can find a lot of variants reported, with 4 being pathogenic. However, pharmacologically, only one splice mutation reported in the gene has been studied in the African population, concerning RO5263397, a schizophrenia drug, by Fowler et al [21]. The study shows reduced UGT2B10 glucuronidation activity due to an alternate splice site, resulting in incomplete mRNA formation and thus reduced activity.
Amine oxidases
Amine oxidases are involved in the oxidative deamination of amines to form corresponding carbonyl derivatives, ammonia and hydrogen peroxide [22].
The metabolism of levocetirizine, here being the only SGA requiring amine oxidases, has not been studied vis-à-vis which specific member of the superfamily is showing the enzymatic action.
Cholyl-CoA-synthetase
Cholyl-CoA-synthetase is involved in taurine conjugation and glycine conjugation in the human body. Better known as Cholate-CoA ligase, this enzyme has the magnesium ion involved as a prosthetic group.
No polymorphism studies as a function of genetics have been conducted on this enzyme.
Sulfotransferases
Sulfotransferases (SULTs) are enzymes catalyzing the transfer of a sulphate group from 3-phosphoadenlyl sulphate to the acceptor specie.
Over the past three decades, studies have been conducted into the overall superfamily, concerning genetic polymorphisms. However, if we consider them concerning mizolastine, the SGA which shows extensive hepatic sulphation, we find that the specific isoenzymes involved in the metabolism of mizolastine have not been studied enough to correlate genomic data with the metabolism.
For a broad understanding of the SULT polymorphism, one can refer to the paper by Kurogi [23].
GSH-S-Transferases
The function of GSH-S-Transferases is primarily considered as the detoxification of the body from xenobiotics by deactivation by glutathione conjugation. Many oxygenating species produced in the body and many xenobiotics use this as the primary detoxifying pathway.
The genetic polymorphisms associated with the GST superfamily have been correlated to many neurological dysfunctions mainly. However, metabolism-specific studies have been conducted by Hayes which show that certain allelic variants show null activity [24,25].
Elimination
With respect to elimination, most SGAs discussed are eliminated by the renal and biliary pathways, passively, without the use of any pathways. Fexofenadine shows an active release into the intestinal tract via P-glycoprotein, as attributed to its accumulation when St. John’s Worts is ingested [26].
A direct clinical correlation between fexofenadine and the P-gp, encoded by the MDR1 gene and associated SNPs has not been and variability in the MDR1 gene was discussed by Yan-Hong Li et al. [27].
Pharmacodynamics
The pharmacodynamics of a drug is a function of how well they interact with its target receptor. In the case of SGAs, the receptor is the H1 receptor. The H1 receptor is a G-protein coupled receptor (Gq). The mode of action of the H1 receptor is through the NF-κB transcription factor [28].
The efficacy of a drug is a function of its affinity to the receptor and the concentration around said receptor.
• In a comparative study between desloratadine, levocetirizine and fexofenadine, the order of affinity stood as desloratadine>levocetirizine>fexofenadine at physiological pH [29].
• The resulting study, after accounting for concentration, placed desloratadine as the most effective, then levocetirizine and then fexofenadine. The criteria were receptor occupancy, for which the values were 95%, 90% and 71% respectively.
Discussion
Another factor which can influence the pharmacodynamics of SGAs is any variability in the H1 receptor. The H1 receptor gene is without any introns and is located on chromosome 3. It has no reported splice variants.
If we discuss the polymorphisms associated with the said gene, we find that there have been studies and papers associating the allelic variants of this gene with unexpected clinical findings. J Li, found more severe side effects in Chinese patients after desloratadine treatment [30]. Jin-Tau Chu found varying efficacies of oral H1 antihistamines with varying genotypes in the Chinese Han population [31].
However, we have not been able to conclusively find which allele is specific for which SGA and which allele is, irrespective of epigenetic factors, showing the highest pharmacodynamic action with the current set of SGAs.
Conclusion
Over the course of this paper, we discussed how, at every step post entry of an SGA into our system, our genetic makeup affects how well it works and for how long it works in the therapeutic range of dose. We first discussed the variability of plasma proteins when we considered the distribution aspect of pharmacokinetics. We then discussed the variability of metabolizing enzymes from the P450 cytochrome superfamily, specifically CYP3A4, CYP2C9, CYP2C19 and CYP2D6. We then went on to discuss the UGT superfamily, followed by GSHS- transferases, amine oxidases and sulfotransferases. A reference to cholyl-CoA-synthetase was also made. We then went on to elimination, specifically talking about P-gp and fexofenadine. We finally went to the pharmacodynamic side and discussed the HRH1.
If we consider and wish to prescribe a specific SGA for anyone, a consideration of all the above factors must be taken. Because we are a function of our DNA.
Conflict of Interests
None stated.
Funding and Disclosures
None.
References
- Tripathi KD. Essentials of medical pharmacology. 7th ed, Jaypee Brothers Medical Publishers, New Delhi, India. 2013:173.
- Johansson A, Enroth S, Palmblad M, Deelder AM, Bergquist J, Gyllensten U. Identification of genetic variants influencing the human plasma proteome. Proc Natl Acad Sci. 2013;110(12):4673-4678.
- Matsumoto S, Yamazoe Y. Involvement of multiple human cytochromes P450 in the liver microsomal metabolism of astemizole and a comparison with terfenadine. Br J Clin Pharmacol. 2001;51(2): 133-142.
- Phase IV Results of Bepreve™, submitted to the FDA by ISTA Pharmaceuticals, Inc. 022288_Bepotastine_Clinpharm_PREA (fda.gov).
- Ketotifen, DrugBank: Ketotifen: Uses, Interactions, Mechanism of Action | DrugBank Online.
- Benedetti MS, Plisnier M, Kaise J, Maier L, Baltes E, Arendt C, el at. Absorption, distribution, metabolism and excretion of (14C) levocetirizine, the R enantiomer of cetirizine, in healthy volunteers. Eur J Clin Pharmacol. 2001;57(8):571-582.
- Wilson CO, Gisvold O. Textbook of organic medicinal and pharmaceutical chemistry. 11th Ed, Lippincott Williams and Wilkins, Pennsylvania, United States. 2004;85-121.
- Falany CN, Johnson MR, Barnes S, Diasio RB. Glycine and taurine conjugation of bile acids by a single enzyme. Molecular cloning and expression of human liver bile acid CoA: Amino acid N-acyltransferase. J Biol Chem. 1994;269(30):19375-193759.
- Lebrun-Vignes B, Diquet B, Chosidow O. Clinical pharmacokinetics of mizolastine. Clin Pharmacokinet. 2001;40(7):501-507.
- Rupatadine, Drugbank: Rupatadine: Uses, Interactions, Mechanism of Action | DrugBank Online.
- Klein K, Zanger UM. Pharmacogenomics of cytochrome p450 3a4: Recent progress toward the "missing heritability" problem. Front Genet. 2013;4:12.
- Ozdemir V, Kalow W, Tang BK, Paterson AD, Walker SE, Endrenyi L, et al. Evaluation of the genetic component of variability in CYP3A4 activity: a repeated drug administration method. Pharmacogenetics. 2000;10(5):373-388.
- Rodríguez-Antona C, Sayi JG, Gustafsson LL, Bertilsson L, Ingelman-Sundberg M. Phenotype-genotype variability in the human CYP3A locus as assessed by the probe drug quinine and analyses of variant CYP3A4 alleles. Biochem Biophys Res Commun. 2005;338(1):299-305.
- Klein K, Thomas M, Winter S, Nussler AK, Niemi M, Schwab M, et al. PPARA: A novel genetic determinant of CYP3A4 in vitro and in vivo. Clin Pharmacol Ther. 2012;91(6):1044-1052.
- Cavallari LH, Momary KM. Chapter 6-Pharmacogenetics in cardiovascular diseases. Pharmacogenomics (Second Edition), Academic Press. 2019:133-179.
- Li‐Wan‐Po A, Girard T, Farndon P, Cooley C, Lithgow J. Pharmacogenetics of CYP2C19: functional and clinical implications of a new variant CYP2C19*17. Br J Clin Pharmacol. 2010;69(3): 222-230.
- Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: Part I. Clin Pharmacokinet. 2009;48(11):689-723.
- Miners JO, McKinnon RA, Mackenzie PI. Genetic polymorphisms of UDP-glucuronosyltransferases and their functional significance. Toxicology. 2002;181-182:453-456.
- Iwai M, Maruo Y, Ito M, Yamamoto K, Sato H, Takeuchi Y. Six novel UDP-glucuronosyltransferase (UGT1A3) polymorphisms with varying activity. J Hum Genet. 2004;49(3):123-128.
- Ehmer U, Vogel A, Schütte JK, Krone B, Manns MP, Strassburg CP. Variation of hepatic glucuronidation: Novel functional polymorphisms of the UDP-glucuronosyltransferase UGT1A4. Hepatology. 2004;39(4):970-977.
- Fowler S, Kletzl H, Finel M, Manevski N, Schmid P, Tuerck D, et al. A UGT2B10 splicing polymorphism common in African populations may greatly increase drug exposure. J Pharmacol Exp Ther. 2015;352(2):358-367.
- Mondovì B, Agrò AF. Structure and Function of Amine Oxidases. In: Bossa, F., Chiancone, E., Finazzi-Agrò, A., Strom, R. (eds) Structure and Function Relationships in Biochemical Systems. Adv Exp Med Biol. 1982;148:141-153.
- Kurogi K, Rasool MI, Alherz FA, El Daibani AA, Bairam AF, Abunnaja MS, et al. SULT genetic polymorphisms: physiological, pharmacological and clinical implications. Expert Opin Drug Metab Toxicol. 2021;17(7):767-784.
- Hayes JD, Strange RC. Glutathione S-transferase polymorphisms and their biological consequences. Pharmacology. 2000;61(3):154-166.
- Dasari S, Gonuguntla S, Ganjayi MS, Bukke S, Sreenivasulu B, Meriga B. Genetic polymorphism of glutathione S-transferases: Relevance to neurological disorders. Pathophysiology. 2018;25(4):285-292.
- Church MK, Church DS. Pharmacology of antihistamines. Indian J Dermatol. 2013;58(3):219-224.
- Li YH, Wang YH, Li Y, Yang L. MDR1 gene polymorphisms and clinical relevance. Yi Chuan Xue Bao. 2006;33(2):93-104.
- Canonica GW, Blaiss M. Antihistaminic, anti-inflammatory and antiallergic properties of the no sedating second-generation antihistamine desloratadine: a review of the evidence. World Allergy Organ J. 2011;4(2):47-53.
- Church DS, Church MK. Pharmacology of antihistamines. World Allergy Organ J. 2011;4(3):S22–S27.
- Li J, Chen W, Peng C, Zhu W, Liu Z, Zhang W, et al. Human H1 receptor (HRH1) gene polymorphism is associated with the severity of side effects after desloratadine treatment in Chinese patients with chronic spontaneous uticaria. Pharmacogenomics J. 2020;20(1):87-93.
- Chu JT. Histamine H1 receptor gene polymorphism acts as a biological indicator of the prediction of therapeutic efficacy in patients with allergic rhinitis in the Chinese Han population. J Cell Biochem. 2019;120(1):164-170.
Citation: Singh A (2023) Pharmacogenetics of Second Generation Antihistamines: A Systematic Review. J Pharmacogenom Pharmacoproteomics. 14:31.
Copyright: © 2023 Singh A. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.