
Indexed In
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- JournalTOCs
- The Global Impact Factor (GIF)
- China National Knowledge Infrastructure (CNKI)
- Ulrich's Periodicals Directory
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2013) Volume 4, Issue 2
Development of Celecoxib Complexes: Characterization and Cytotoxicity Studies in MCF-7
Abstract
The objective of the study was to compare the complexing ability of Celecoxib with different complexing agents. Inclusion complex of Celecoxib is well established in literature but its evaluation in terms of thermodynamics was not studied elaborately. Hence after characterization of complexes with Differential scanning calorimeter, X-ray diffraction, NMR and % complexation efficiency, its solvation energetics and thermodynamics were determined. Complexation ability of celecoxib with caffeine was also evaluated, where non-inclusion (charge transfer) mechanism is involved. So, these two mechanisms of complexation were also tested with respect to each other. In this manuscript, new complexing agents (humic acid and fulvic acid) were also introduced for Celecoxib. These lie in the category of natural organic matter and we explored an indigenous source (Shilajit) to extract it. Other agents used for the development of complex were HP-β-CD and β-CD. Release mechanism of drug from complexes was studied by in vitro release studies. MTT assays were also performed to assess in vitro cell toxicity potential of the complexes in comparision to neat drug.Introduction
Over the past decade, significant advances have been made in understanding the molecular and cellular events that regulate and mediate angiogenesis [1]. The great interest in vascular biology and angiogenesis research is mainly driven by the potential for therapeutic interventions in angiogenesis-dependent diseases such as cancer and chronic [2]. Non-steroidal anti-inflammatory drugs (NSAIDs) and selective COX-2 inhibitors are widely prescribed drugs for the treatment of pain and inflammation. They inhibit the catalytic activity of the cyclooxygenase isoenzymes and thereby block the production of prostaglandins and thromboxanes, the two main classes of lipidderived pro-inflammatory molecules. Increasing evidence suggests that NSAIDs and COXIBs might also prove efficient anti-angiogenic drugs [3-5].
It is much understood that solid tumor growth is dependent upon the growth of new blood vessels which is known to under the heading of an angiogenesis [6]. Initially for tumorogenesis multiple studies have been demonstrated that the degree of tumor vascularity core corresponds positively with disease stage i.e., metastases, and cancer recurrence. Angiogenesis also plays a master role in hematogenous malignancies, such as leukemia, lymphoma, and multiple myeloma, as well as in premalignant myelodysplastic syndromes. Antiangiogenic therapy has been developed and validated as an effective cancer treatment strategy for a growing number of cancer types including colorectal, renal, liver, lung, brain, pancreatic, neuroendocrine tumors (NET), gastrointestinal stromal tumors (GIST), multiple myeloma and myelodysplastic syndrome. More than 120 new antiangiogenic entities are in clinical, trials [7]. Finally, preclinical, and clinical data is demonstrating that angiogenesis inhibition can be applied for achieving chemoprevention studies [8].
Celecoxib, 4-[5-(4-methyl phenyl)-3-(triflouromethyl)-1Hpyrazol- 1-yl], is a non-steroidal anti-inflammatory drug (NSAID), which is a specific cyclooxygenase-2 (COX-2) inhibitor and is estabilished as an anti-angiogeneic agent. The major drawback with celecoxib therapy is its poor aqueous solubility and dissolution in gastric fluid. Hence it is aimed to enhance the aqueous solubility and dissolution rate of celecoxib by forming inclusion complexes with different complexing agents (Figure 1) like humic acid (HA), fulvic acid (FA), β-cyclodextrin (β-CD), 2-hydroxypropyl-β-cyclodextrin (HP- β-CD) and caffeine (Caff). Newly introduced humic acid and fulvic acid belong to natural organic matter which form inclusion complex with therapeutic molecules [9]. Cyclodextrins are torusshaped oligosaccharides obtained from the enzymatic degradation of starch by bacteria [10], and are known to form inclusion complexes with many lipophilic organic molecules both in solution and in the solid-state. Caffeine is a natural xanthine alkaloid compound that acts as a central nervous system stimulant which is reported to form complex by a charge transfer mechanism [11]. Various celecoxib complexes were formulated and their physiochemical properties were investigated. These were then evaluated with a through comparision of different techniques. The complex which performed appreciably better is purported for future formulation development.

Figure 1: Molecular structures of celecoxib (CXB) and complexing agents.
Materials and Methods
Rock shilajit was obtained and authenticated from Dabur research foundation, Ghaziabad, India. A slightly modified method [9] was used to extract FA and HA from rock shilajit. Celecoxib (COX-2 inhibitor) was a gift sample by IPCA Pharmaceutical Ltd., Mumbai, India. Chemicals and reagents used for the study were of A.R. grade and purchased from Merck, Mumbai, India.
Equilibrium phase solubility studies
Phase solubility studies were carried out at room temperature (25°C) in triplicate according to the method reported by Higuchi and Conners. Excess amount of CXB was added to distilled water containing various concentrations (0.2-2% w/v) of different complexing agents in a series of stoppered conical flasks (100 ml) and shaken for 48 h on a rotary flask shaker. The suspensions were filtered by membrane filter paper (0.45 μm). The solubility of CXB was assessed by a slight modification of a validated HPLC method [12], with an accuracy of 96.5% and a precision of 6.4%. The analysis was carried out on a Waters Alliance e2695 separating module (Waters Co., MA, USA) using photodiode array detector (Waters 2998) with auto sampler and column oven. The instrument was controlled by use of Empower software installed with the equipment for data collection and acquisition. Compounds were separated using a C18 reverse phase column (25×4.6 mm, particle size 5 μm, Merck, Germany) maintained at room temperature. The mobile phase consisted of methanol–water, 75:25 (%, v/v). The flow rate was 1 ml/min and the detection wavelength was 252 nm. 10 μL of the sample was injected into the column. Rt of the CXB was observed at 5.2 min [13].
Preparation of complex by Freeze drying method
The inclusion complexes in the molar ratio of 1:1 quantity of drug was added to aqueous solutions of the complexing agents and were agitated on a magnetic stirrer for 24 h. Sucrose solution was (2% w/v) added as a cryoprotectant. The resulting solutions were frozen at (-70°C) in a deep freezer for overnight. This was then lyophilized (Dry Winner, DW-8-85 Heto Holten, Denmark). The resulting mass was then powdered in a glass mortar and pestle and passed through a 100-mesh sieve to obtain a uniform-size fine powder. The samples were then transferred into a vacuum desiccator and dried over silica gel under vacuum for atleast 24 h. A similar batch of complex, without cryoprotectant was also developed to carry out instrumental analysis like DSC and XRD.
Characterization of complexes
Following methodologies were employed to characterize the complexes.
Morphology of the complex: Morphology of the inclusion complex was studied using scanning electron microscopy (SEM). For SEM analysis (SEM, Zeiss Instrument), a drop of complex suspension was mounted on aluminium stub with the help of black carbon tape and air dried at room temperature. Next, the samples were placed in a vacuum chamber of SEM gold coating apparatus and gold was coated at 2.5 kv, 25-30 mA for 180 seconds. The morphology of the particles using SEM figures 2a and 2b at 25.0 kv, nominal magnification of 13,000–40,000 and scan speed=8 were observed.

Figure 2: Morphology of optimized inclusion complex.
Differential scanning calorimeter: DSC thermograms were obtained under a nitrogen gas flow of 50 ml/min. Calibration of the DSC instrument (DSC-7, Perkin Elmer Pyris 6 instrument, USA) was carried out using indium as a standard. Sample powders (5 mg) were crimped in an aluminum pan and heated at a rate of 10 K min−1. Generally, scan rates are taken between 1 and 10 K/min. Low scan rates are preferable in terms of peak resolution and investigation of the sample having close peaks while high scan rates increase the sensitivity of the measurement as they lead to the exchange of heat within a comparatively short time period. Further, scan rate may also influence the course of temperature related processes within the sample. Samples were heated from 30 to 350°C (highest melting points of organic substances).
X-ray diffraction: XRD samples were studied using X-ray diffractometer (PW 1830, Phillips, Banglore, Karnataka, India). The samples (100 mg) on XRD plates were rotated during data collection to reduce orientation effects of particles. XRD patterns of all the samples were recorded between 2θ=5° and 70° at 35 kV and 30 mA, respectively.
Proton nuclear magnetic resonance (1H NMR): Proton Magnetic Resonance (1H NMR) spectra were recorded on Brucker Model DRX- 300 NMR spectrometer in CDCl3 using tetra methyl silane (TMS) as the internal standard. Chemical shifts are reported in parts per million (ppm, δ) and signals are described as a single (s), doublet (d), triplet (t), quartet (q) and multiple (m).
Determination of complexation efficiency: The ability of different agents to complex celecoxib was determined quantitatively by UV spectrophotometry in triplicate mode. Acetonitrile was used for the determination of free CXB content since complexing agents taken, had poor solubility in acetonitrile and the inclusion complex was also expected to have low solubility in acetonitrile. To estimate free CXB content, 10 mg of the sample was dissolved in 100 ml of acetonitrile. This solution was then sonicated for 2 min, filtered on a 0.45 μm filter and analyzed using UV spectroscopy (UV 1601, Shimadzu, Japan) at 256 nm. The free CXB content was determined in triplicate. Following equation was used to determine the complexation efficiency:
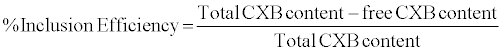 (1)
(1)
Saturation solubility (aqueous) of complexes: Excess amount of different complexes were added to 10 ml of an aqueous solution (pH 7.1 ± 0.2). They were shaken in a water bath (Ray Scientifics Instruments, India) for 5 days at 37°C, centrifuged (Tomy MX-305, Japan) at 3000 g for 10 min and filtered. The concentration of CXB in the resulting solutions was then analyzed by the method described in methodology section. The resulting values were the average of at least three replicates.
The standard solution Gibbs energies were calculated using following equation:
ΔG°sol=−RTlnX2
Where, X2 is the molar fraction of CXB in a saturated solution.
The standard solution enthalpies were derived from temperature dependencies of drug solubilities expressed in molar fractions (van’t Hoff equation):
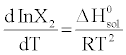 (2)
(2)
For use of the above equation following assumptions were made:(a) the activity coefficients of dissolved drugs do not deviate from unity and (b) the solution enthalpies do not depend on concentration. The solution heat capacities are considered to be constant within studied temperature range, since the temperature dependence of solubility is described by linear equations.
Thermodynamics of inclusion complexes: The thermodynamic parameters (ΔH, ΔS and ΔG) for the formation of inclusion complex were determined from temperature dependence of apparent association constants, by using classical van’t Hoff equation, and plotting ln K versus 1/T. The corresponding enthalpy and entropy can be obtained from the slope and intercept, respectively.
In vitro release study
In vitro release study of active pharmaceutical ingredient (100 mg CXB suspension) and inclusion complexes (equivalent to 100 mg CXB) was performed using USP II dissolution apparatus (Hanson Research SRS, Chatsworth, CA, USA) in 900 ml of double distilled water containing 1% sodium lauryl sulphate at 50 rpm and 37° ± 2°C. The concentration of CXB was determined using HPLC method described. The study was carried out by transferring the constituted suspension (5 ml) in dialysis bag (Spectra-Por dialysis bag supplied by Sigma Aldrich, St. Louis, MO, USA with cutoff 12,000–14,000 Da). All dissolution studies were carried out in triplicate.
In vitro cell cytotoxicity studies of complexes
An MTT assay was used to assess the cytotoxicity of the free drug as well as the control and the complexes. The MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) assay is based on conversion of yellow water soluble tetrazolium dye to a waterinsoluble purple formazan by living cells. The amount of formazan generated is directly proportional to the number of viable cells.
MCF-7 cells (ATCC, USA grown in NII, New Delhi) were grown (37°C, 5% CO2 in water jacketed incubator shell) using DMEM media (Dulbeccos modified eagle medium for cell culture growtrh, Gibco Invitrogen, USA) with 10% FBS (Foetal bovine serum) and seeded on a single 96 well plate (Corning costar, USA) and allowed to adhere for MTT assays. Following this a concentration of free drug as well as the control and complexes ranging from 50 μg/ml to 500 μg/ml having equivalent concentration were added in duplicates to the single 96 well tissue culture plate (Falcon Plate, Corning costar, USA). After 24 hours of treatment, the MTT assay was performed to check cell viability [14]. For the MTT assay, the media was removed from all the wells, 10 μl of MTT reagent (Chemicon International, USA) per well from a working stock (5 mg/ml) was added and the plates incubated (37°C and 5% CO2) for 2-3 hrs, after that the reagent was removed and the crystals were solubilised using isopropyl alcohol (IPA). This IPA extract (Isopropanol with 0.1 N HCL) was then transferred to 96 wellplates. The HCl converts the phenol red in the tissue culture medium to a yellow colour that does not interfere with the MTT formazan measurement. The Isopropanol dissolves the formazan to give a homogeneous blue solution. The absorbance was measured at a test wavelength of 570 nm and reference wavelength of 630 nm using an ELISA plate reader, LMR-340 M (Labexim International, Austria). The value of absorbance is a measure of the number of live cells.
Results and Discussion
Equilibrium phase solubility studies
Phase solubility analysis of CXB in presence of different complexing agents are given in figure 3. Here different natures of interaction between complexing agents and CXB have been observed. Basically, two types of interactions were observed. In one category curve fitting line was inclined towards Y-axis while in other category, it was inclined towards X-axis. This tendency may be used to predict the comparative orders of complexation. With respect to complexing agents, HA and FA were exhibiting lower order of complexations as compared to HP β CD, CD and Caffeine. The total solubility (STotal) of a drug as a function of concentration of different complexing agents is given by any of the following equations which also give an estimate to predict the stoichiometry of the complexation. For single order complexation, solubility relationship is expressed by:

Figure 3: Phase solubility studies at room temperature (25°C) in triplicate mode.
STotal=S0+m [DmCA]
Where, S0 is the drug concentration is the absence of complexing agents and m refers to the stoichiometry of the complexation. Here, one molecule of the drug interacts with one molecule of complexing agents. Thus, solubility of CXB can be assumed linear and proportional with complexing agent when stoichiometry of complexation is 1. This is the case with HA, FA and others. For second order complexation (one drug molecule interacts with two molecules of complexing agents), fitting the data to a quadratic model is appropriate. It is expressed by:
STotal=S0+K1:1S0[CA]+K1:1K1:2S0[CA]2
A positive deviation from the linearity for the phase-solubility diagram and deconvolution of the curve is observed with higher order complexes. So except caffeine (1:2) all other complexing agents appear to follow first order with respect to complexing agents.
A phase solubility graph is also used to determine the binding constant and other thermodynamic (ΔG, ΔH and ΔS) parameters [15]. The binding constant was calculated according to the formula:
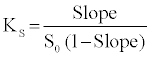 (3)
(3)
Where, S0 is the solubility of CXB without complexing agents.
The binding constant for different complexes was found to be varying like CXB-FA (1216.84 M-1), CXB-HA (996.54 M-1), CXB-HP- β-CD (654.29 M-1), CXB- β-CD (481.78 M-1) and CXB- Caffeine (448.54 M-1). Stability constant (Ks) values lying between 200 and 5,000 M-1 are considered as most suitable for the improvement of solubility and stability of a poorly soluble drug [16]. To investigate the complexation process in the prespective of thermodynamics, Van`t Hoff equation was used. A plot is drawn between 1/T and Log K and thermodynamics drawn is discussed.
Characterization of complexes
Morphological analysis: Optimized inclusion-complex suspensions were analysed using SEM and are given in figures 2a and 2b. SEM analysis of complex is helpful to characterize the size and morphology of the particles. SEM analysis of complex revealed that the inclusion complex were spherical shape with a smooth surface.
Differential scanning calorimeter: DSC is a tool to investigate the melting and recrystallization behavior of crystalline materials. Organic substances usually show a melting range. An increased melting range could be correlated with impurities or less ordered crystals. The DSC analysis showed a sharp peak of CXB at 156.812°C. None of the complexing agents showed any sharp peak except Caff at 194°C. All the complexes exhibited blunt humps at variable temperature ranges as shown in figure 4. It may be inferred from the thermograms that CXB may have lost its crystalline pattern and thus is present in its amorphous form. The details from the thermograms of CXB and its complexes are given in table 1.
| Samples | ΔH (j/g) | Maximum peak (°C) | Peak width |
|---|---|---|---|
| FA | 142.075 | 162.387 | 11.13 |
| HA | 44.236 | 77.365 | 44.63 |
| HP-β-CD | 33.768 | 119.876 | 31.42 |
| β-CD | 117.96 | 127.625 | 41.26 |
| Caffeine | 124.630 | 194.524 | 7.46 |
| CXB | 163.671 | 156.812 | 17.23 |
| CXB- Caff | 143.261 | 156.253 & 191.452 | 12.56 |
| CXB- β-CD | 53.624 | 153.264 | 9.45 |
| CXB - HP-β-CD | 74.364 | 161.261 | 11.39 |
| CXB-HA | -- | -- | -- |
| CXB -FA | 33.211 | 101.624 | 5.64 |
Table 1: Differential scanning calorimteric data of pure Celecoxib, complexing agents and complexes. Peak width was calculated manually.

Figure 4: Representative differential scanning calorimetry profiles of different samples.
Melting enthalpy (ΔH), expressed in joule/gram (J/g) is a characteristic of the crystal order if the influence of impurities can be ignored. For the less ordered crystal or amorphous state, the melting of the substance requires less energy than the perfect crystalline substance which is required to overcome the lattice energy. As a result, the higher melting enthalpy values should suggest higher ordered lattice arrangement and vice versa. The higher enthalpies with single entities like CXB (163.671 j/g), FA (142.075 j/g), β-CD (117.96 j/g) and Caffeine (124.630 j/g) were noted. So far the complexes are concerned, ΔH values were comparatively lower. Thus, it may be inferred from the results that complexes have more disordered structures. This decrease in disorder created due to positive interaction between two entities (drug and complexing agents).
Peak width is another parameter to study but it depends largely upon particle size especially in nanometric range as explained by Gibbs–Thomson equation:
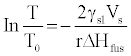 (4)
(4)
Where, T=Melting temperature of a particle with radius r
T0=Melting temperature of the bulk material at the same external pressure
γsl=Interfacial tension at the solid–liquid interface
Vs=Specific volume of the solid
ΔHfus=Specific heat of fusion
Peak width is actually an integration of peaks of different size ranges. When the particle size distribution is wide spread, it is higher and vice versa. The particle size of unprocessed drug is around 148.3 nm with a PDI of 0.6. Other samples were passed through sieve 100 hence; a higher size distribution could be expected.
Although DSC is able to monitor and quantify even minute thermal events in the sample (depending on the sensitivity of the instrument) and to identify the temperatures at which these events occur. But it does not directly reveal the cause of a thermal event. The exact nature of the thermal transitions has to be determined with other complementary methods such as microscopic observations,
X-ray diffraction or spectroscopic techniques to distinguish, between melting, polymorphic transitions, loss of water from hydrates and decomposition of the substance.
X ray diffraction: Use of these two techniques (DSC and XRD) often lead to complementary information on the systems of interest and data evaluation from these methods is usually straightforward. Such a plot (2θ vs. % Intensity) can be considered a fingerprint of the crystal structure and to differentiate with different crystallographic status of the system. One peak will be exhibited for all repeating planes with the same spacing. By contrast, an amorphous sample will exhibit a broad hump in the pattern called an amorphous halo. These patterns are representative of the structure but do not give positional information about the atoms in the molecule. Celecoxib was showing a characteristic crystalline pattern in figure 5 exhibiting intense peaks at 22.36 (100%), 17.33 (97.59%), 05.36 (96.53%), 12.34 (88.53%), 30.32 (16.66%) and 41.5 (11.9%) etc. Few sharp peaks were present only in caffeine (100% at 11.24, 49.56% at 27.32, and 47.64% at 28.42) and HP-β-CD (100% at 12.34, 39.40% at 23.68, 42.52% at 25.34 etc). Other complexing agents (FA, HA and β-CD) were almost amorphous in nature are represented in figure 6. XRD of CXB-HA and CXB-FA showed in figure 5, amorphous nature and some diminished peaks were observed in CXB- HP-β-CD, CXB-β-CD and CXB- Caffeine complexes. These peaks were not from the fingerprint regions of CXB but from the complexing agents itself. Thus, the XRD of inclusion complexes showed their amorphous nature, indicating entrapment of drug and, peaks if any were appreciably diminished.

Figure 5: X-ray diffraction of complexes.

Figure 6: X-ray diffraction of celecoxib and complexing agents.
Proton nuclear magnetic resonance (1H NMR): The 1H NMR spectra of CXB in figure 7 showed one singlet at δ 7.15 due to pyrazole proton. While 1H NMR spectra of CXB-β-CD complex showed singlet peaks at δ 7.19. All the other peaks of both β-CD and CXB remain almost unchanged. This clearly indicates the inclusion phenomena of the pyrazole nucleus into the β-CD cavity [17]. Nearly same phenomena was observed for inclusion complex of CXB with FA, HA and HP- β-CD. The downfield shift values observed were δ 7.22 for CXB-FA, δ 7.17 for CXB-HA, δ 7.23 for CXB- HP- β-CD. 1H NMR spectra of CXBcaffeine complex also showed changes in both the protons and shifting pattern of the proton in CXB-and caffeine. H8 proton of caffeine was observed at δ 7.90 where as CXB-caffeine complex showed H8 peak as upfielded doublet at δ 7.80.

Figure 7: NMR spectroscopy of celecoxib.
Determination of complexation efficiency: The results obtained for complexation efficiency studies are given in table 2. Except CXBCaffeine complex (~46.54%) all other complexation was appreciable. In author`s point of view this may not be a very appropriate method to determine the complexation efficiency. High sonication energy may result into dissociation of no bond complexes (inclusion complex) since with small gastric movement we expect the release of molecule from the inclusion complex. However, due to unavailibility of any appropriate method, authors had to follow the reported method.
| Samples | % Complexation efficiency ± SD (n=3) |
|---|---|
| 1:1 FA-CXB | 72.32 ± 2.1 |
| 1:1 HA-CXB | 61.11 ± 0.9 |
| 1:1 CXB- HP-β-CD | 64.48 ± 2.3 |
| 1:1 CXB- β-CD | 59.33 ± 2.4 |
| 1:2 CXB- Caff | 46.54 ± 3.8 |
Table 2: Complexation efficiencies of different complexes. Inclusion of drug appears to be a better approach than charge-transfer mechanism to associate a drug with an inert excipient.
Saturation solubility (aqueous) of complexes: The maximum solubility was observed with 1:1 CXB-FA complex (73.4 μg/ml). Decreasing order of solubility is given in table 3. Thermodynamic parameters (ΔG, ΔH and ΔS) calculated for the samples are also given in the table 4. Positive signs of enthalpy of solublisation (ΔH) indicate that it is an endothermic process at room temperature and crystal lattice energy of the substance overweighs the solvation energy. Positive values of ΔG also indicate that the process is non-spontaneous at room temperature and is driven by some energy source. Nature of change of disorder (ΔS) of the system is different (negative) in all the systems (complexes). Since disorder of the system is decreasing we may expect that both the components (drug and complexing agents) are obtaining an ordered structure after associating with each other.
| Sample | Aqueous solubility (µg/ml) | Mole fraction | ΔG298sol (kJ mol−1) | ΔH298sol (kJ mol−1) | T ΔS298 sol (kJ mol−1) | ΔS298sol (J mol−1 K−1) |
|---|---|---|---|---|---|---|
| CXB | 6.4 | 0.302´10−6 | 37.18 | 47.1 ± 1.3 | -9.92 | -33.2886 |
| CXB-FA | 73.4 | 3.464´10−6 | 31.15 | 48.2 ± 1.4 | -17.05 | -57.2148 |
| CXB-HA | 61.6 | 2.907´10−6 | 31.58 | 39.1 ± 0.1 | -7.52 | -25.2349 |
| 1: 1 CXB-β CD | 43.8 | 2.067´10−6 | 32.42 | 38.3 ± 0.5 | -5.88 | -19.7315 |
| CXB-HP- β- CD | 44.9 | 2.11´10−6 | 32.37 | 39.6 ± 0.2 | -7.23 | -24.2617 |
| CXB- Caff | 32.7 | 1.543´10−6 | 33.15 | 49.4 ± 1.1 | -16.25 | -54.5302 |
Table 3: Saturation solubility of complexes at pH 7 (aqueous). Experiment was carried out at room temperature (298 K) and in a triplicate mode.
| Parameters | CXB-β-CD | CXB-HP-β-CD | CXB-FA | CXB-HA | CXB-Caff |
|---|---|---|---|---|---|
| ΔH (KJ mol−1) | -16.11 | 1.52 | 1.59 | 3.17 | 7.62 |
| ΔG (KJ mo−1) | -16.34 | -0.259 | -0.364 | 1.426 | 6.576 |
| ΔS (J mol–1 K−1) | 0.769 | 5.97 | 6.56 | 5.849 | 3.501 |
| T ΔS (J mol–1 K−1) | 0.229 | 1.779 | 1.954 | 1.743 | 1.043 |
Table 4: Association constants and thermodynamic parameters for complexation of celecoxib with different complexing agents.
Thermodynamics of inclusion complexes: The driving force for complexation was variable for different complexes (Table 4). It was inferred from the change in numerical values of the different variables (ΔH, ΔG and TΔS). CXB-β-CD complex appears to be driven by Gibb’s free energy (-16.34). CXB-HP-β-CD, CXB-FA and CXB-HA by ΔS (5.97, 6.56 and 5.849 respectively). While CXB-Caff complexation appears to be an enthalpy driven (7.62 KJ mol–1) process. Except CXB- β-CD, formation of all the complexes was endothermic in nature (+ve; ΔH) at room temperature which further translates into the fact that crystal lattice energy of the substance outweighs the solvation energy.In the table 4, negative value of ΔG is related with spontaneity of the process while positive values of ΔG indicate that the process is nonspontaneous at room temperature and is driven by some energy source.
In vitro release study
The results obtained after dissolution study are given in figure 8. To analyze the in vitro release data various kinetic models were used to describe the release kinetics. The zero order release describes the systems where the drug release rate is independent of its concentration. The first order describes the release from system where release rate is concentration dependent (order is one). Higuchi described the release of drugs from insoluble matrix as a square root of time dependent process based on Fickian diffusion. Whereas, Peppas Korsemeyer describes the log fraction of drug release with respect to log time. Release order of all the complexes appears to be first order (Table 5).
| Formulations | Zero-order | First-order | Higuchi matrix | Korsmeyer-peppas | Best fit model | |
|---|---|---|---|---|---|---|
| r2 | r2 | r2 | r2 | Rel. Expo. (n) | ||
| CXB-FA | 0.967 | 0.995 | 0.959 | 0.960 | 0.326 | First order |
| CXB- HA | 0.967 | 0.995 | 0.959 | 0.960 | 0.326 | First order |
| CXB- HP-β-CD | 0.967 | 0.995 | 0.959 | 0.960 | 0.326 | First order |
| CXB-β-CD | 0.967 | 0.995 | 0.959 | 0.960 | 0.326 | First order |
| CXB-Caff | 0.967 | 0.995 | 0.959 | 0.960 | 0.326 | First order |
Table 5: Release kinetics and mechanism involved with different complexes. Release pattern with highest r2 value is used to predict the kinetics involved.

Figure 8: In vitro release profile of different complexes.
To evaluate the mechanism of drug release from the preparation, data of drug release may be plotted in Korsmeyer and Peppas equation,
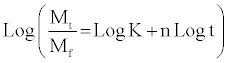 (5)
(5)
Where Mt is the amount of drug release at time t, Mf is the amount of drug release after infinite time; k is a release rate constant incorporating structural and geometric characteristics of the dosage form, n is the diffusional exponent indicative of the mechanism of drug release. The log value of percent drug dissolved is plotted against log time for each formulation according to the equation. For a cylinder shaped matrix the value of n ≤ 0.45 indicates Fickian release; >0.45 but <0.89 for non- Fickian (anomalous) release; and >0.89 indicates super diffusion type of release. It generally refers to the erosion of the polymeric chain and anomalous transport (Non-Fickian) refers to a combination of both diffusion and erosion controlled drug release. In all five complexes release appears to be Fickian diffusion (n=0.326).
In vitro cytoxicity studies of complexes
The corresponding values for optical density for the control, neat drug and the complexes are shown in figure 9. The CXB was found not to have cytotoxic effect. At the lowest concentration (50 μg/ml) relative difference of cell viability was not distinguishable with each other, although they were not interfering with cell growth. Some negative patterns of β-CD complexes in cell growth was observed at higher concentrations (100 μg/ml, 200 μg/ml and 500 μg/ml) as compared to other complexing agents. However, for the complexes the toxicity did not increase at different concentrations. So far the humic substances are concerned; they are also reported to be safe in cytotoxicity studies [18]. Thus, the reports of in vitro cell toxicity study did not indicate any cellular toxicity.

Figure 9: MTT assay results of pure drug and complexes.
Conclusion
Celecoxib-inclusion complexes have been successfully used to improve the performance of this poorly soluble drug. Numerous complexing agent along with two natural Humic acid and Fulvic acid, a naturally occurring substance, has oriented its suitability as a complexing agent which can successfully developed the potential of celecoxib as an antiangiogenic agent can be used as an adjuvant for chemoprevention in MCF-7. Lastly, the data revealed the importance of inclusion complex in improving the solubility of the complex and its eventual use in development of different types of formulations.
References
- Carmeliet P (2000) Mechanisms of angiogenesis and arteriogenesis. Nat Med 6: 389-395
- Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1: 27-31.
- Gately S (2000) The contributions of cyclooxygenase-2 to tumor angiogenesis. Cancer Metastasis Rev 19: 19-27.
- Cavallaro U, Christofori G (2000) Molecular mechanisms of tumor angiogenesis and tumor progression. J Neurooncol 50: 63-70.
- Langer K, Balthasar S, Vogel V, Dinauer N, von Briesen H, et al. (2003) Optimization of the preparation process for human serum albumin (HSA) nanoparticles. Int J of Pharm 257: 169-180.
- Folkman J (2001) Angiogenesis-dependent diseases. Semin Oncol 28: 536-542.
- Li WW, Hutnik M (2007) Angiogenesis-based medicine: principles and practice for disease prevention and intervention. Angiogenesis: Basic Science and Clinical Applications.
- Albini A, Noonan DM, Ferrari N (2007) Molecular pathways for cancer angioprevention. Clin Cancer Res 13: 4320–4325.
- Mirza MA, Agarwal SP, Rahman MA, Rauf A, Ahmad N, et al. (2011) Role of humic acid on oral drug delivery of an antiepileptic drug. Drug Dev Ind Pharm 37: 310-319.
- Szetjli J (1998) Introduction and general overview of cyclodextrin chemistry. Chem Rev 98: 1743–1753.
- Fouad EA, El-Badry M, Alanazi FK, Arafah MM, Al-ashban R, et al. (2012) Preparation and investigation of acetyl salicylic acid-caffeine complex for rectal administration. Pharm. Dev Tech 15: 249-257.
- Baboota S, Faiyaz S, Ahuja A, Ali J, Shafiq S, et al. (2007) Development and Validation of A Stability-Indicating HPLC Method For Analysis of Celecoxib (CXB) In Bulk Drug and Microemulsion Formulations. Acta Chromatographica 18: 116-129.
- Hazem A, Hassan, Ali H, Alaa A, Hamza, et al. (2007) Enhancement of dissolution amount and in vivo bioavailability of itraconazole by complexation with ß-cyclodextrin using supercritical carbon dioxide. J Pharm Biomed Anal 45: 243–250.
- Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65: 55-63.
- Yadav VR, Suresh S, Devi K, Yadav S (2009) Effect of cyclodextrin complexation of curcumin on its solubility and antiangiogenic and anti-inflammatory activity in rat colitis model. AAPS Pharm Sci Tech 10: 752–762.
- Patel RP, Patel MM (2007) Preparation and evaluation of inclusion complex of lipid lowering drug lovastatin with ß-cyclodextrin. Dhaka Univ J Pharm Sci 6: 25-36.
- Spulber M, Pinteala M, Fifere A, Harabagiu V, Simionescu B (2008) Inclusion complexes of 5-flucytosine with ß-cyclodextrin and hydroxypropyl- ß-cyclodextrin: characterization in aqueous solution and in solid state. J Incl Phenom and Macrocycl Chem 62: 117-125.
- Yamada P, Isoda H, Han JK, Talorete TP, Abe Y (2007) Inhibitory effect of fulvic acid extracted from Canadian sphagnum peat on chemical mediator release by RBL-2H3 and KU812 cells. Biosci Biotechnol Biochem 71: 1294-1305.
Copyright: © 2013 Alam MA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.