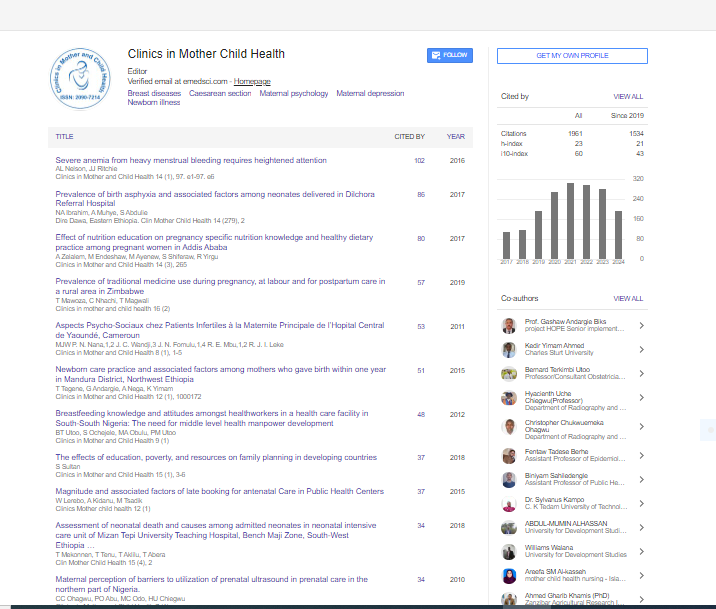

Indexed In
- Genamics JournalSeek
- RefSeek
- Hamdard University
- EBSCO A-Z
- Publons
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2020) Volume 17, Issue 4
Caroli Syndrome Associated with Polycystic Kidney and Congenital Heart Disease in Children in Africa
Sow A1*, Boiro D1, Gueye M1, Ndongo AA2, Sow PS1, Dieye S1, Ba A3, Ba I3, Thiongane A3, Keita Y2, Dieng YJ3, Deme I3, Niang B3, Sow NF3, Seck MA3, Fatah M3, Diagne G3, Mbaye A3, Houngbadji M3, Kane A3, Bop K3, Coundoul AM3, Mbodj M3, Ndiaye ST3, Cisse DF3, Faye PM3 and Ndiaye O32Department of Pediatrics, Aristide le Dantec Hospital Center, Dakar, Senegal
3Department of Pediatrics, Albert Royer National Children Hospital, Dakar, Senegal
Received: 15-May-2020 Published: 15-Jun-2020, DOI: 10.35248/2090-7214.20.17.351
Abstract
Caroli syndrome (CS) is defined as the association of Caroli disease with congenital liver fibrosis. It is a rare congenital pathology with a global prevalence of less than 1 / 1,000,000 inhabitants. We report a case of CS associated with polycystic kidney disease (PKD). He is a 7 years old boy followed from the age of 2 years for epigastric pain. Abdominal ultrasounds were performed at the ages of 2 years, 4 years and 6 years. They showed hepatomegaly associated with dilation of the intrahepatic bile ducts, hepatic fibrosis and bilateral renal cysts and micro-stones. The child received several hospitalizations, the last of which was at the age of 6 years for abdominal pain, ascites, digestive haemorrhage secondary to portal hypertension and hematuria. On admission, the weight was 19 kg (-2 DS), the size was 119cm (-1 DS) and the BMI was 13.41 (-3DS). The child had no icterus. The abdomen was distended and a painless hepatomegaly was palpated with a hepatic arrow at 12.5cm and stage II splenomegaly. The urine test showed hematuria. The liver test showed an isolated elevation of alkaline phosphatase. The abdominal computed tomography scan showed hepatomegaly, dilation of the intrahepatic bile ducts and the presence of bilateral renal hypodense cystic lesions. The patient received Aldactone, Propanolol, Captopril, a blood transfusion and ursodesoxycholic acid as treatment. The evolution is stationary awaiting surgical management.
Keywords
Caroli; Heart; kidney; child; Senegal
Introduction
Caroli's disease (CD) is a congenital pathology characterized by segmental dilation of the intrahepatic bile ducts. Caroli syndrome (CS) is defined by the association of CD with congenital hepatic fibrosis [1, 2]. CS and CD are rare and the global prevalence is less than 1 / 1,000,000 inhabitants worldwide [2]. In Africa, a few cases of CS have been described. In Senegal, only one case of CD was described in 2016 [3]. It is in this context that we report a rare case of Caroli syndrome associated with polycystic kidney disease (PKD) in a young boy in Senegal.
Observation
He is a 7 years and 4 months old boy. Fetal ultrasonography during pregnancy were normal. Toxoplasmosis, rubella, human immunodeficiency virus (HIV), syphilis and hepatic serologies were negative. At birth, he weighed 2850g, his height was 52cm and his head circumference was 36cm. The child has benefited from exclusive breastfeeding until 4 months before a food diversification with cereals and vegetables. He has normal psychomotor development. The parents have no link of inbreeding and they are healthy . The patient is the only child of the couple. The child has a heterozygous sickle cell profile (AS). At the age of 2 years 6 months, a moderate congenital mitral insufficiency without repercussions on the cardiac chambers was detected (Figure 1).

Figure 1.Heart doppler ultrasound showing moderate congenital mitral regurgitation
The boy has been followed since the age of 2 years and 6 months for recurrent epigastric pain which motivated several consultations and investigations. Abdominal ultrasounds were performed at the ages of 2 years, 4 years and 6 years. They showed hepatomegaly associated with segmental dilation of the intrahepatic bile ducts, hepatic fibrosis and bilateral renal cysts and micro-stones. The results of the ultrasounds are illustrated in (Figure 2).

Figure 2. Abdominal ultrasound showing hepatomegaly with cystic dilation of the intrahepatic bile ducts and bilateral renal microstones
The child received several hospitalizations, the last of which was at the age of 6 years for portal hypertension syndrome. He presented abdominal pain, ascites, digestive hemorrhages by rupture of esophageal varices type 2. This symptomatology was associated with hematuria. At admission, he had a weight of 19 kg (-2 DS), a height of 119cm (-1 DS), a BMI of 13.41 (-3DS), a respiratory rate at 15 cycles / min, a heart rate at 98 beats / min, blood pressure 80 / 70mmhg and Spo2 at 99%. Consciousness was normal. The child was pale without jaundice. There were no edemas in the lower limbs. At heart level there was a breath of mitral insufficiency of intensity 3/6.
The abdomen was distended. There was hepatomegaly, painless, of regular surface and firm consistency. The hepatic arrow was estimated at 12.5 cm. There was also a stage II splenomegaly of the HACKET classification. Biological analyzes noted an anemia at 8 g / dl normochromic normocytic regenerative. The rest of the hemogram was normal. Cytobacteriological examination of urine (ECBU) was normal. The urinary tests showed hematuria at 21,750 red cells / min and leukocyturia at 62,250 leukocytes / min. The results for liver and kidney function are shown in (Table 1).
| Liver function tests | Parameters | Results | Normal |
|---|---|---|---|
| AST | 45UI/l | (<37UI/l) | |
| ALT | 23UI/l | (<23UI/l) | |
| Ɣ-GT | 41UI/l | (<35UI/l) | |
| Alkaline phosphatase | 473 UI/l | (98-279UI/l) | |
| Bilirubinemie totale | 8.6mg/l | (3-10mg/l) | |
| Bilirubinemie directe | 2.17mg/l | (<5mg/l) | |
| Prothrombin | 97.3% | (>70%) | |
| urea | 0.23g/l | (0,15-0,39 g/l) | |
| Kidney fucntion tests | creatinemia | 09mg/l | (27-45mg/l) |
| Uric acid | 47,6 mg/l | (35-72mg/l) | |
| Calcemia | 100mg/l | (90-107mg/l) | |
| Phosphoremia | 43,2 mg/l | (27-45mg/l) | |
| Blood sodium | 137,7 mEq/l | (135-145mEq/l) | |
| Blood potassium | 3,97 mEq/l | (3,5-5,5 mEq/l) | |
| Urinary Sodium | 55 mmol/24h | (40-220mmol/24h) | |
| Urinary Potassium | 21 mmol/24h | (23-123mmol/24h) | |
| Urinary calcium | 28mg/24h | (100-300 mg/24h) | |
| Phosphaturia | 430mg/24h | (400-1300mg/24h) | |
| Oxaluria | 83umol/24h | (80-490 umol/24h) | |
| cystinuria | 120mg/24h | (<400mg/24h) |
Table 1 : Results of biological examinations of the patient's hepatic and renal function
An abdominal computed tomography scan showed hepatomegaly with dilation of the intrahepatic bile ducts in continuity with a cystic formation of 3cm in lobe V with bilateral renal hypodense cystic lesions (Figure 3).

Figure 3. Abdominal computed tomography (CT) showing cystic dilation of the intrahepatic bile ducts with bilateral renal hypodense lesions
The diagnosis was Caroli syndrome (SC) associated with polycystic kidney disease. The genetic study was not done. The patient received aldactone, propanolol, captopril, blood transfusion and ursodeoxycholic acid as treatment. The evolution is stable pending surgical management (ligation of esophageal varicose vein and hepatic surgery).
Discussion
SC is a congenital pathology but the diagnosis is often late [4]. A study in Morocco reported an average age at diagnosis of 38 years while another multicenter study found an average age of 53 years [4, 5]. The delay in diagnosis is explained by an insidious course of the disease with nonspecific symptoms. The disease often appears at the stage of complications such as portal hypertension [4]. In our patient, the symptomatology started at the age of 2 years 6 months and the diagnosis was made at the age of 7 years 4 months which is relatively early compared to the data in the literature.
This rapid development in our patient could be explained by the presence of comorbidities such as congenital heart disease, kidney disease and hemoglobinopathy. The most commonly described clinical sign is epigastric pain or pain in the right hypochondrium. Our patient has had abdominal pain since the age of 2 years. This symptomatology is classically described in the literature [6]. Examination at the stage of complications often identifies a portal hypertension syndrome with ascites, splenomegaly and gastric haemorrhage without jaundice [4].These same clinical signs were found in our patient. Laboratory tests to explore liver function is often normal.
Most often only an elevation of alkaline phosphatase (ALP) is noted [7]. Our patient has high ALP (> 2 x normal). Transaminases and bilirubinemia were normal. The diagnosis is based on imaging including ultrasound, computed tomography scan (CT), magnetic resonance imaging (MRI) of the bile ducts and cholangiopancreatography [2]. The best examination is currently the MRI of the bile ducts which has a better diagnostic sensitivity [4]. Liver biopsy is rarely necessary in the diagnosis [2]. However MRI of the bile ducts is not very accessible and very expensive in our contexts. In our patient the diagnosis was made on the basis of abdominal ultrasound and CT. The association of CS with polycystic kidney disease is frequent because it involves the same genes.
CS is most often associated with autosomal recessive polycystic kidney disease (AKAR) while autosomal dominant polycystic kidney disease (ADPK) is most often associated with hepatic cysts [8]. Kidney damage is often asymptomatic for a long time [8]. In our patient, only one episode of hematuria was noted since the onset of symptoms, but we note the presence of ultrasound renal micro-stones. However, regular monitoring is necessary since there is a risk of chronic renal failure in adulthood. We did not do the genetic study because it is not very accessible in our contexts. The association of SC and congenital heart disease has not been described in the literature to our knowledge. congenital heart disease affecting the mitral valve is rare [9]. Our patient has mitral insufficiency without severe repercussions on heart function. Medical treatment of SC is based on beta-blockers and ursodeoxycholic acid, however the best treatment is surgery or liver transplantation [5].
Conclusions
CS is a rare pathology and often late diagnosis. The evolution in children can be rapid towards complications such as portal hypertension, especially in cases of associated pathology such as PKR and heart disease. The diagnosis can be strongly guided in our contexts by ultrasound and CT.
REFERENCES
- Caroli J. Diseases of the intrahepatic biliary. Clin Gastroenterol. 1973;2:147-161.
- Yadav P, Adhikari S, Pandit N, Awale L, Vasan K, Khadka S. Caroli’s disease: a diagnostic challenge. Int Surg J. 2018;5(11):3750-3753.
- Boiro D, Gueye M, Dieng YJ, Ndongo AA, Thiongane A, Seck N, et al. Maladie de Caroli à révélation précoce : A propos d’un cas au service de néonatologie du Centre Hospitalier Abass Ndao de Dakar. Méd Afr Noire. 2016;63(11) :553-558.
- Kettabi F, Benzzoubeir N, Errabih I, Ouazzani L, Ouazzani H. Maladie et syndrome de caroli : deux entites rares (à propos de 5 cas). Journal Marocain des Sciences Médicales 2017;21 (1):20-25.
- Mabrut JY, Partensky C, Jaeck D, Oussoultzoglou E, Baulieux J, Boillot O, et al. Congenital Intrahepatic Bile Duct Dilatation is a Potentially Curable Disease: Long-Term Results of a Multi-institutional Study. Ann Surg. 2007;246(2): 236-245.
- Madjov R, Chervenkov P, Madjova V, Balev B. Caroli’s disease. Report of 5 cases and review of literature. Hepatogastroenterology 2005;52:606-609.
- Suchy FJ. Caroli's disease. 2006.
- Shenoy P, Zaki SA, Shanbag P, Bhongade S. Caroli’s Syndrome with Autosomal Recessive Polycystic Kidney Disease. Saudi J Kidney Dis Transpl. 2014;25(4):840-843.
- Carpentier A, Branchini B, Cour JC, Asfaou E, Villani M, Deloche A. Congenital malformations of the mitral valve in children: Pathology and surgical treatment. J Thorac Cardiovasc Surg.1976; 72(6):854-866.
Citation: Sow A, Boiro D, Gueye M, Ndongo AA, Sow PS, Dieye S, et al. (2020) Caroli Syndrome Associated with Polycystic Kidney and Congenital Heart Disease in Children in Africa. Clinics Mother Child Health. 17:351. DOI: 10.35248/2090-7214.20.17.351.
Copyright: © 2020 Sow A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.