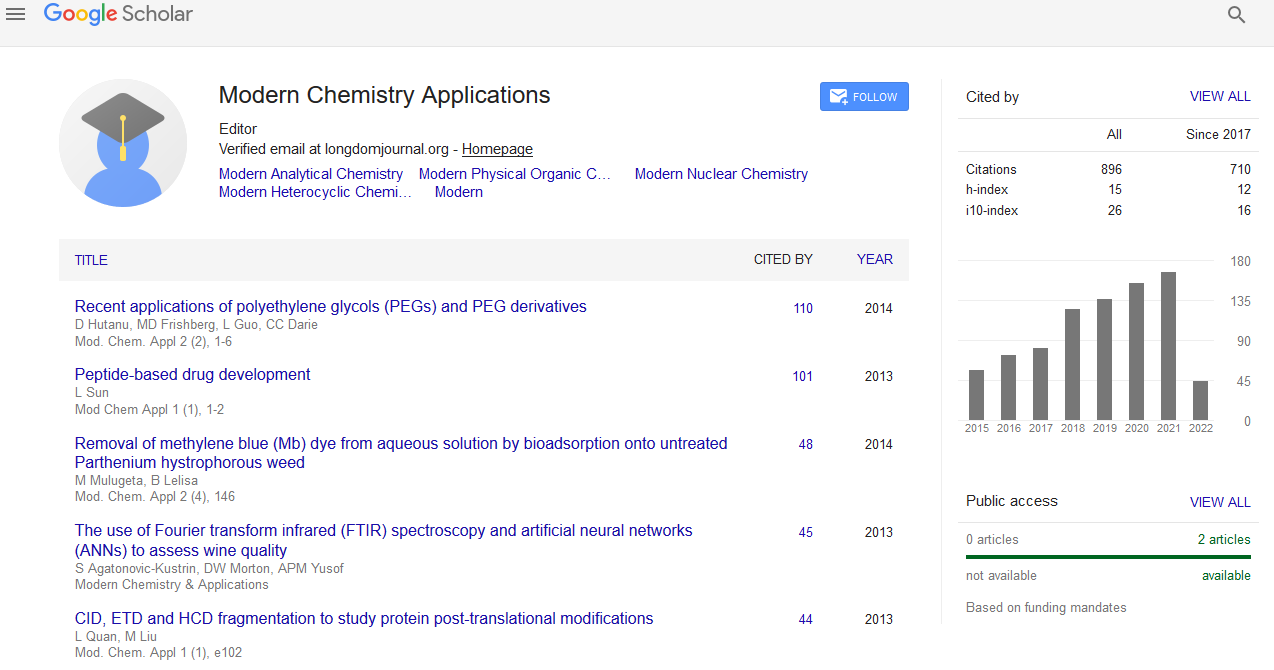

Indexed In
- Open J Gate
- JournalTOCs
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Scholarsteer
- Publons
- Geneva Foundation for Medical Education and Research
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Commentary - (2025) Volume 13, Issue 1
Progress in Computational Chemistry for Predictive Modeling and Rational Molecular Design
Emili Davson*Received: 26-Feb-2025, Manuscript No. MCA-25-29065; Editor assigned: 28-Feb-2025, Pre QC No. MCA-25-29065 (PQ); Reviewed: 14-Mar-2025, QC No. MCA-25-29065; Revised: 21-Mar-2025, Manuscript No. MCA-25-29065 (R); Published: 28-Mar-2025, DOI: 10.35248/2157-7560.25.13.493
Description
The development of computational chemistry has significantly influenced how researchers understand, predict and design chemical systems. Over recent years, the field has matured with increased computing resources, refined algorithms and more accurate theoretical models. These improvements have helped move beyond traditional trial-and-error approaches, enabling scientists to estimate properties and behavior of molecules before entering the laboratory.
One important contribution of computational chemistry is its ability to predict molecular structures and reactivities. Quantum mechanical methods, particularly density functional theory, are now widely used to calculate electronic distributions, molecular orbitals and potential energy surfaces. These calculations are helpful for interpreting experimental observations and evaluating chemical stability. As algorithms become more efficient, it is possible to study larger systems and more complex interactions, making the models more relevant for real-world applications.
Another valuable aspect is the simulation of dynamic behavior. Molecular dynamics allows researchers to observe how atoms and molecules move over time under different conditions. This approach can reveal information about temperature-dependent properties, conformational changes and reaction pathways. Coupling these simulations with machine learning has added new dimensions to the field. Algorithms can be trained on large datasets to predict trends, improving accuracy and saving computational time.
The use of data-driven models has grown quickly. Machine learning tools are being applied to screen compound libraries, predict binding affinities and estimate physicochemical properties such as solubility, toxicity and absorption. These methods are especially useful in early stages of drug discovery, where thousands of potential compounds must be evaluated. By filtering candidates through computational predictions, resources can be directed toward the most likely successful options.
In materials science, simulations assist in understanding interactions at surfaces, interfaces and within bulk phases. These insights help in designing materials with desirable electrical, thermal and mechanical properties. For instance, computational models have been used to predict how molecules interact with electrode surfaces in batteries or how catalysts behave under operational conditions. Such applications guide experimental work and reduce the time needed to identify useful compounds.
Modeling is also being used in understanding biological systems. Protein-ligand interactions, enzymatic reactions and membrane dynamics are studied through a combination of structural bioinformatics, docking and free energy calculations. These efforts support the development of new treatments and allow for exploration of mechanisms that may be difficult to capture experimentally.
The development of new functionals, basis sets and algorithms continues to improve accuracy. However, each method has limitations and choosing the right approach depends on the size and nature of the system being studied. Computational chemists must balance speed, accuracy and available resources when selecting tools for their work.
Integration with experimental data is another area where computational chemistry excels. It provides a framework for testing hypotheses, suggesting reaction conditions and interpreting results. For example, infrared or NMR spectra can be simulated and compared with experimental ones to validate structures. Similarly, reaction mechanisms proposed from experimental evidence can be evaluated by calculating transition states and energy barriers.
The increased accessibility of software and databases has made these methods available to a wider community. Open-source tools, cloud-based platforms and shared datasets enable collaboration and reproducibility. This openness helps in refining models, verifying results and building upon previous work, thereby making progress more reliable and efficient.
Educational efforts have also contributed to the spread of computational methods. As more institutions include these topics in their curriculum, a new generation of chemists becomes familiar with both the theory and application of modeling techniques. This knowledge helps integrate computational thinking into research and development workflows across academia and industry.
Overall, computational chemistry continues to expand the possibilities for chemical research and application. With the ability to test ideas in silico, researchers are able to evaluate numerous possibilities quickly, explore theoretical systems and gain a deeper understanding of chemical behavior. As computing power and algorithms continue to advance, the influence of this field will likely become even more integrated into everyday scientific practice.
Citation: Davson E (2025). Progress in Computational Chemistry for Predictive Modeling and Rational Molecular Design. Modern Chem Appl. 13:493.
Copyright: © 2025 Davson E. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.