
Indexed In
- Academic Journals Database
- Open J Gate
- Genamics JournalSeek
- JournalTOCs
- China National Knowledge Infrastructure (CNKI)
- Scimago
- Ulrich's Periodicals Directory
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- MIAR
- University Grants Commission
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2021) Volume 12, Issue 3
Insights of How Lung Microbiome can Contribute to COVID-19 Severity in Intensive Care Unit Patients
Fabíola Marques de Carvalho1, Leandro Nascimento Lemos1, Luciane Prioli Ciapina1, Rennan Garcias Moreira2, Alexandra Gerber1, Ana Paula C. Guimarães1, Tatiani Fereguetti3, Virgínia Antunes de Andrade Zambelli3, Renata Avila3, Tailah Bernardo de Almeida4, Jheimson da Silva Lima5, Shana Priscila C. Barroso6, Mauro Martins Teixeira7, Renan Pedra Souza8, Cynthia Chester Cardoso9, Renato Santana Aguiar2 and Ana Tereza R. de Vasconcelos1*2Department of General Biology, Institute of Biological Sciences, Federal University of Minas Gerais, Belo Horizonte, Brazil
3Department of Infectology, Eduardo de Menezes Hospital, Belo Horizonte, Brazil
4Department of Biology, Almirante Paulo Moreira Sea Studies Intitute, Brazilian Navy, Rio de Janeiro, Brazil
5Department of Burns Treatment, Marcílio Dias Navy Hospital, Rio de Janeiro, Brazil
6Department of Molecular Biology, Biomedical Research Institute, Marcílio Dias Navy Hospital, Rio de Janeiro, Brazil
7Department of Biochemistry and Immunology, Institute of Biological Sciences, Federal University of Minas Gerais, Belo Horizonte, Brazil
8Department of Genetics, Ecology and Evolution, Institute of Biological Sciences, Federal University of Minas Gerais, Belo Horizonte, Brazil
9Department of Genetics, Biology Institute, Instituto de Biologia, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
Received: 28-Apr-2021 Published: 20-May-2021, DOI: 10.35248/2157-7560.21.12.450
Abstract
Objectives: Secondary bacterial and fungal infections are associated with respiratory viral infections and invasive mechanical ventilation. Microbiome influence on COVID-19 severity in patients admitted to intensive care units (ICU) remains poorly understood. This work described the lung microbiota of Brazilian COVID-19 patients and explored how microbial pathogens can contribute to Coronavirus disease 2019 clinical outcome.
Methods: Total DNA of bronchoalveolar lavage fluids from 21 Brazilian COVID-19 patients was extracted. All patients were positive RT-PCR and admitted to intensive care units in two Brazilian centers. For metagenomic analyses, sequenced reads were submitted to bioinformatic tools for taxonomic and functional inferences.
Results: We identified respiratory, nosocomial, and opportunistic pathogens as prevalent bacteria in the lung, suggesting a dysbiosis process (microbial imbalance) in ICU COVID-19 patients. Microbial functional analyses showed metabolic pathways associated with virulence repertoire, such as biofilm production, secreted toxins, capsular polysaccharides, and iron acquisition. Microbial pathogens and their virulence mechanisms were associated with host immunological responses, and a cellular model suggesting how bacterial species could participate in COVID-19 worsening was presented.
Conclusion: We explore how microbial species present in the lung could potentially modulate and aggravate the immunological processes of patients admitted to intensive care units, contributing to COVID-19 severity.
Keywords
COVID-19, Respiratory tract infections; Metagenomics; Bacteria; Metabolism; Bioinformatics
Introduction
Severe COVID-19 cases are characterized by systemic hyperinflammation and immune dysfunction, leading to lung pathophysiological effects that include edema, endothelial and epithelial injuries, vasoconstriction, inflammation, and fibrosis [1]. An influx of neutrophils into the interstitium and bronchoalveolar space results in impairment of arterial oxygenation, which together with infection of alveolar macrophages by SARS-CoV, trigger the cytokine storm [2]. In the critical stage of COVID-19, cytokine storm contributes to vascular congestion, complement cascade activation, and over disseminated intravascular coagulation, having an extensive role in COVID-19 severity and mortality [3,4].
Hypoxia is aggravated, and invasive mechanical ventilation may be required for life maintenance. Under conditions of oxygen tension variation, alveolar ventilation, blood flow, and concentration of inflammatory cells, microbial growth conditions in the lung are directly affected [5,6]. Irregular innate immune response in acute lung injury and neutrophil recruitment can favor bacterial infections, which are also regulated by virulence factors [7].
Secondary bacterial infection during Intensive Care Unit (ICU) admission and use of invasive mechanical ventilation was reported in Influenza and Severe Acute Respiratory Syndrome (SARS). Data in literature pointed out coinfection or secondary bacterial pneumonia observed in 11% to 35% of individuals with laboratory confirmed influenza [8]. Furthermore, Streptococcus pneumoniae and Staphylococcus aureus occurred in 35% and 28% of patients, respectively. In Canada, Chlamydophila pneumoniae or Mycoplasma pneumoniae were abundant in up to 30% of SARS patients [9]. In pulmonary and catheter-associated infections, A. baumannii, Klebsiella pneumoniae, and Enterococcus faecium are species frequently recovered from endotracheal tubes [10-12].
Bacterial infection was also identified in COVID-19 cases [13- 15]. Studies in the USA, China, Spain, and Thailand, showed secondary bacterial infection present in 14% of patients, with the occurrence of Streptococcus pneumoniae, Klebsiella pneumoniae, and Haemophilus influenzae within 1–4 days of COVID-19 disease onset [16,17]. According to Rawson et al. [18], the extensive use of broadspectrum antibiotics can predispose COVID-19 patients to acquire bacterial infections and increase multidrug resistance.
In Brazil, one of the countries with the highest growth rate in the number of daily deaths and continuous spread of Sars-CoV-2, a recent study of composition data of 250,000 hospital admissions for COVID-19 revealed that in-hospital mortality among patients admitted to the ICU was 59% and among those who received mechanical ventilation was 80%, with patients aged ≥60 years remaining as the majority cases group [19]. Despite this, knowledge about colonizing species in patients infected with SARS-CoV-2, the prevalence of bacterial infection, and the mechanism of co- or secondary infection are still poorly understood. Advances on clinical microbiology have been expanded by metagenomic approaches that allow a full spectrum of host-microbiome by identifying species without the need for culture-based methods [20]. An investigation focused on bacterial microbiota in Brazilian SARS-CoV-2 cases was conducted in only one patient. The concise results showed the predominance of Lautropia, Prevotella, and Haemophilus genera. However, an in-depth work was not performed [21].
Here we performed a metagenomic study on 21 Brazilian COVID-19 patients to describe their microbiota and investigated the functional role of these species on the Coronavirus disease 2019. The study aimed to bring to light how microbial pathogens could potentially contribute to the immunological responses characteristic of the disease and the clinical outcome of patients admitted to intensive care units.
Materials and Methods
Study cohort and data collection
The study included 21 COVID-19 patients admitted to Intensive Care Units (ICU) from two metropolitan cities from the Southeast Brazilian region (from Marcílio Dias Navy Hospital, Rio de Janeiro, and Eduardo Menezes Hospital, Minas Gerais). Samples were obtained in different periods of pandemic (from May 7 to September 17, 2020). Although more invasive and difficult to collect to the detriment of blood and gut samples, bronchoalveolar lavage fluids were selected to better reflect the pulmonary microbiota. SARS-CoV-2 patients were confirmed based on RT-PCR tests. The present study was conducted considering communityacquired COVID-19 positive and symptomatic patients. Samples identification was modified to preserve the patient's anonymity. The Ethics Commission from Marcílio Dias Navy Hospital and Eduardo Menezes Hospital approved the present study (protocol number 32382820.3.0000.5256 and 31462820.3.0000.5149). Characteristics of the 21 patients were obtained from medical records and are summarized in Supplementary Table 1.
DNA extraction and sequencing
DNA from bronchoalveolar lavage fluids samples from hospitalized patients was extracted using standard manufacturer's protocols for the Qiagen QIAamp DNA Microbiome Kit (Qiagen, Germany). Metagenomic libraries were constructed using the Nextera DNA Flex Library Preparation Kit (Illumina, USA). Sequencing was performed in a NextSeq 500 System with a NextSeq 500/550 High Output Kit v2.5 (300 Cycles) (Illumina, USA).
Bioinformatics analysis
Sequencing files were submitted to MG-RAST (version 4.0.3) [22] for taxonomical and functional inferences. Sequences were trimmed using default parameters, and human sequences were removed by screening against Homo sapiens. Species were identified from MG-RAST using RefSeq, and functional abundances were obtained using the SEED subsystem database. For both analyses, the Best Hit Classification criteria and alignment cutoff applied included an e-value>10-5, a minimum identity of 60%, and a minimum alignment of 15. Phyloseq R package was used to estimate the Shannon diversity index [23]. Comparative analyses were conducted considering normalized data. Variations in sampling depth were adjusted by random subsampling implemented in Phyloseq R until all samples had the same library size. Microbiome R package was applied to detect the core microbiome [24], using as a cutoff criterion species that occurred in more than 75% of patients. Correlation levels between most abundant enzymes and species previously identified were done using the Corrplot R package [25], applying Pearson method and FDR (false discovery rate) adjusted-P<0.05 for statistical significance. Correlation scale varied from -1 (negative correlation) to 1 (positive correlation). Scatter plots were generated using ggplot2 R [26].
Results
Clinical aspects of COVID-19 patients
Sampling comprised cases occurred in May (the third month of the pandemic in Brazil) (47.62%), June-July (months of COVID-19 wide spreading and peak) (42.85%), and August-September (months of stability or reduction stages in the number of cases in Brazil) (9.53%). The study included SARS-CoV-2 individuals confirmed by RT-PCR and admitted to Intensive Care Units (ICU), which date of hospital discharge or death were available (Supplementary Table 1). Time intervals between the onset of symptoms and sample collection varied from 4 to 34 days. The median age was 62 years [IQR, 50–72]. Patients older than 65 years composed 42.86% of the sampling. Overall, 57.14% of patients were men. Fever and dyspnoea were more frequent clinical symptoms, with oxygen saturation<95% observed in 85.71% of patients. Despite advanced age as a COVID-19 risk factor, pre-existing comorbidities were present in 95.23% of patients, most commonly represented by diabetes (47.62%), obesity or hypertension (33%), and cardiovascular disease (23.8%). Invasive ventilatory support was necessary in 95.2% of the cases. No previous pulmonary impairment was reported, except for one individual. Interstitial infiltrates detected by X-ray were observed in 52.38% of cases. Information about antibiotic administration was obtained only for 33% of patients, being linezolid and azithromycin the most common drugs. Dosage and time for the administration of antibiotics were not reported. The hospitalization period varied from 2 to 55 days, and the mortality rate was 71.42% (Supplementary Table 1).
Species diversity on COVID-19 ICU patients and the microbial metabolic mechanisms to evade the host immune response
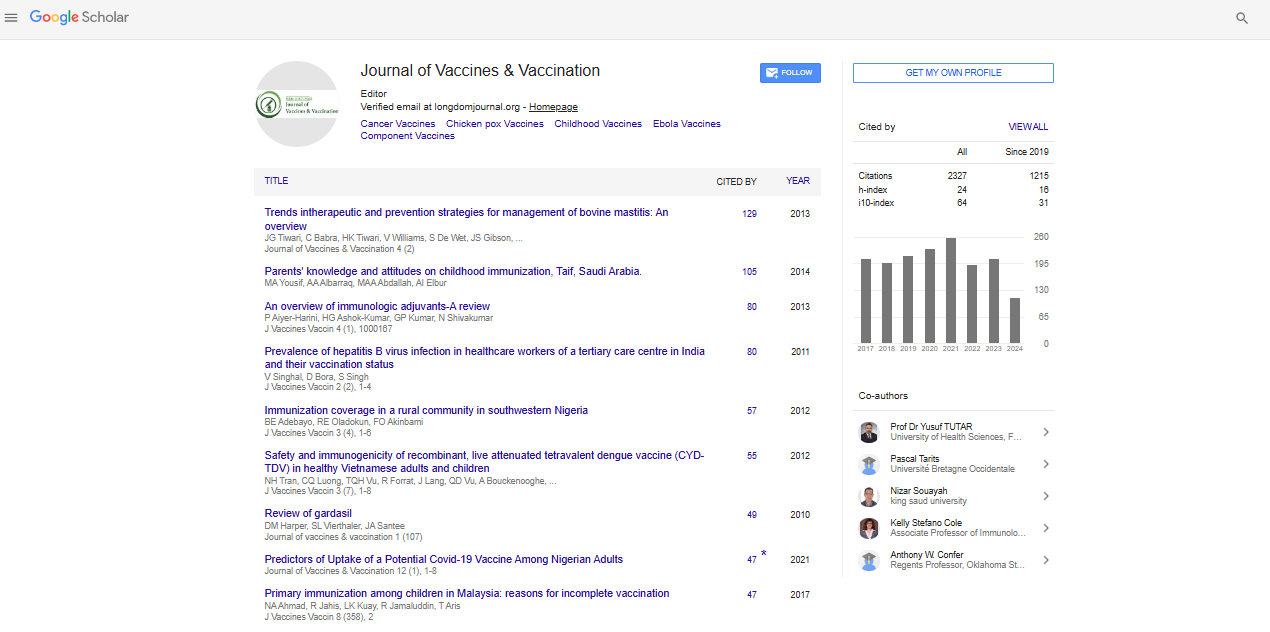
The fifty most prevalent species represented almost 75% of all lung microbial diversity present in patients (Figure 1 and Supplementary Table 2). Respiratory and nosocomial microorganisms overrepresented the microbiota. The most common respiratory pathogens identified were Acinetobacter baumannii (corresponding to 16.5% of all bacterial species), Streptococcus pneumoniae (8.8%), Pseudomonas aeruginosa (3.6%), and Staphylococcus aureus (3.4%). Nosocomial pathogens mainly included fungi Candida albicans and Candida tropicalis (representing 11.2% and 8.1% of Eukarya domain), as well as the bacteria Enterococcus faecalis (1.5%) and Escherichia coli (0.5%).

Figure 1: Microbial composition and pathogens prevalence in COVID-19 patients. Relative abundance of the 50 most prevalent species identified in the 21 COVID-19 patients. The characteristics of the individuals analyzed, such as age range, comorbidities, and time (days) from the onset of symptoms until sample collection were represented.
Although less frequently, a diversity of other pathogenic microorganisms was observed, such as the uncommon Listeria monocytogenes, Streptococcus agalactiae, S. pyogenes, sporulating Bacillus (B. anthracis and B. thuringiensis), and Acidovorax spp. (Figure 1). The opportunistic microorganisms Coprobacillus spp. and Delftia acidovorans were present with an abundance of almost 2% and 6%, respectively. Additionally, a large diversity of the oropharynx commensal species was identified, such as Rothia mucilaginosa, Parvimonas micra, Actinomyces odontolyticus, and Streptococcus anginosus (Supplementary Table 2), which can rarely cause respiratory disease in compromised immune individuals.
High occurrence of the bacterial respiratory pathogen Streptococcus pneumoniae was found independent of the comorbidity. The absence of comorbidity was reported only in one patient, which showed a drastic reduction of respiratory and nosocomial pathogens abundance, except for Candida albicans and Candida tropicalis (Figure 1 and Supplementary Table 2). In contrast, Acinetobacter baumannii was significantly increased in patients with the most prolonged periods of SARS-CoV2 symptomatology (samples Mgt-Cov-20,18 days and Mgt-Cov-21, 34 days). No significant difference or direct association was observed between pathogen abundance and comorbidity type.
To check the consequence of pathogens proportion on microbial diversity, we estimated diversity using the Shannon index and calculated the relative abundance of pathogens (Figure 2). The proportion of microbial pathogens was based on the relative abundance of individual pathogens divided by the total number of species in the microbiome. A negative correlation (-0.9660624; p ≤ 0.05) was observed between these two parameters, indicating that the prevalence of pathogens in patients without antibiotic use is inverse to diversity. The high proportion of pathogens leads to a decrease in species richness, which explains the reduced diversity of microorganisms in ICU COVID-19 patients.

Figure 2: Relationship between microbial diversity and pathogens proportion. Pearson correlation between the Shannon diversity index and the relative abundance of pathogenic species identified in those COVID-19 patients without antibiotic administration. A similar pattern was obtained considering all samples.
To understand the functional role of microbial pathogens in COVID-19, the metabolic profile of these microbiota was investigated. Carbohydrate metabolism, amino acid and derivatives and protein metabolism were the most overrepresented categories (Supplementary Table 3). Carbohydrate metabolism was mostly represented by Tricarboxylic Acid (TCA) cycle pathway (18%), with an abundance of the dihydrolipoamide dehydrogenase enzyme, which is associated with virulence in bacteria (Figure 3 and Supplementary Table 4).

Figure 3: Functional profile of microbiome from 21 COVID-19 patients. Metabolic pathways were obtained by MGRAST and analyzed to levels 1 (more generalist categories) until to a more specific level, which identifies enzymes. Most overrepresented categories or enzymes within one specific level are shown.
Within each general category, besides of TCA cycle, the most abundant subpathways included: Type IV secretion system proteins (48%), followed by fatty acid metabolism (40.8%), iron acquisition (40.2%), DNA replication (19.3%), and reactive oxygen species (ROS) protection (18.3%) (Figure 3 and Supplementary Table 1). Type IV secretion system and DNA replication showed proteins related to general DNA metabolism functions. For Protection from Reactive Oxygen Species pathway, catalase was present in abundance, being an active enzyme on the detoxification process of oxidative subproducts (Figure 3 and Supplementary Table 1).
Enoyl-CoA hydratase, an enzyme with participation in biofilm and bacterial resistance, had a dominant frequency within fatty acid metabolism, a pathway whose disorder has a relevant impact on the outcome of some clinical diseases. Additionally, high protein proportion for the iron acquisition category was observed (Figure 3), with the identification of ferrichrome-iron (an iron chelator isolated initially from fungi) and TonB receptor (a membrane localized complex responsible for iron transport across the outer membrane) (Supplementary Table 1).
To obtain a panorama of mechanisms required for microbial evasion and virulence, we investigated proteins of general metabolic profile more deeply in the context of secretion systems; toxins and superantigens, invasion and intracellular resistance, adhesion, and capsular and extracellular polysaccharides pathways. We identified an extensive repertoire of secretion systems (I, II, III, IV, V, VI, VII, and VIII). Virulence factors particularly relevant for bacterialhost interactions were found on secretion systems of Types I and V, highlighting cytolysins toxins (RTX agglutinin and cytolysin activator), proteins involved in heme utilization (exoprotein and hemophore HasA), and adhesin (Figure 4 and Supplementary Table 4).

Figure 4: Metabolic pathways associated with microbial evasion and virulence. Abundant proteins identified on each one of distinct virulence pathways (secretion systems; toxins and super-antigens, invasion and intracellular resistance, adhesion and capsular and extracellular polysaccharides pathways) are shown.
Toxins and superantigens were represented, in a smaller abundance, by export ABC transporter ATP-binding protein and streptolysin proteins (SagI, SagD, and SagH) (Figure 4 and Table 4), suggesting the partial presence of the Streptolysin S locus, which is present in Group A Streptococcus spp. Within the intracellular invasion and host adhesion categories, we found sortase, signal peptidase I, and fibronectin/fibrinogen-binding proteins (Figure 4). In sialic acid metabolism, a useful pathway for bacterial pathogenesis, we observed high proportion of capsular and extracellular polysaccharides, particularly UDP-N-acetylglucosamine 2-epimerase, glucosamine-1-phosphate N-acetyltransferase, and phosphoglucosamine mutase (Figure 4).
Correlation analyses were performed as a function of the association level between the most abundant enzymes and species identified (Figure 5A). We observed a high abundance of dihydrolipoamide dehydrogenase positively correlated to Streptococcus pneumoniae proportion (r=0.834, p-value<0.05). The enoyl-CoA hydratase enzyme of the fatty acid pathway, and the ferrichrome-iron receptor, involved in the iron acquisition were associated with Escherichia coli (r=0.753 and 0.774, respectively,p-value<0.05). Secreted agglutinin RTX, a T1SS toxin, and UDP-N-acetylglucosamine 2-epimerase, a capsular polysaccharide of the sialic acid pathway, had a positive correlation to A. baumannii (r=0.880 and 0.727, respectively, p-value<0.05). Exoprotein involved in heme utilization, adhesion, and virulence was strongly correlated to Pseudomonas aeruginosa (r=0.816, p-value<0.05). Candida albicans showed a weak association to phosphoglucosamine mutase, however without statistical support. Interestingly, the results suggest that even when in low abundance (<1%), E.coli responds in a dependent manner to an increase of abundance enzymes related to fatty acid and iron acquisition pathways (Figure 5A). In addition, when enzymeenzyme correlations were estimated, increase of ferrichrome-iron receptor was accompanied by enoyl-CoA hydratase (r=0.773, p-value<0.05), and UDP-N-acetylglucosamine 2-epimerase showed positive correlation to T1SS secreted agglutinin RTX protein (r=0.607, p-value<0.05) (Figure 5B).

Figure 5A: Correlation among most abundant enzymes-species studied. Estimate of association level among most abundant enzymes and/or species identified in this study. The dependence of one variable was upon another was measured using the relative abundance of a particular species as X-axis value and enzyme proportion as Y-axis value (A).

Figure 5B: Correlation among most abundant enzymes-species studied. Enzyme versus enzyme (B) correlations were also performed. Correlation scale varied from -1 (negative correlation) up to 1 (positive correlation). Only positive and significant associations are shown. Correlation values on scatter plots were obtained by method Pearson at a significance level of 95% confidence. Correlation and p-values indices are represented by r and P, respectively.
Discussion
The microbial pathogens and their virulence mechanisms here reported were associated with immune and clinical disorders of COVID-19, and a cellular scenario in three interconnected stages is presented (Figure 6). First, we showed respiratory pathogens such as Acinetobacter baumannii, Streptococcus pneumoniae, Pseudomonas aeruginosa, Staphylococcus aureus, and Candida spp. present in abundance. The occurrence and the high proportion of respiratory and nosocomial pathogens can be explained by the potential imbalance between microbial immigration and elimination proposed by Dickson and collaborators [6,27]. According to these authors, hyperventilation accelerates the influx of airborne microbes in advanced lung diseases, and microbial elimination is reduced by bronchoconstriction and impaired mucociliary clearance. This condition provides a nutrient-rich growth medium and decreased oxygen concentration, influencing bacterial reproduction.
Second, once bacterial colonization is established, microbial pathogens secrete metabolites to evade the host immune response and enhance their survival (Figure 6). A. baumannii, S. pneumoniae, P. aeruginosa, and S. aureus, may promote virulence and evade the host immune response by biofilm production, induction of cytoor hemo-lysins, iron acquisition factors, cytokines, adhesins, and complement system resistance, besides other virulence factors [28- 30]. Metabolic pathways and enzymes related to these virulence factors were observed in our data.

Figure 6: The interplay of the bacteria found in COVID-19 patients, virulence factors, and free-iron in immune response and disease severity. In severe lung diseases, hyperventilation contributes to the microbial influx and reduces the elimination of microorganisms. Tecidual injury promotes a rich environment for the growth of specific lung pathogens into the alveolar compartment. Bacterial pathogens evade the host immune response by biofilm production, induction of hemolysins, iron acquisition factors, adhesins, and other virulence factors. The SARS-CoV-2 and bacterial pathogens promote erythrocyte lysis, causing heme(Hb)/iron(Fe) liberation. Heme uptake is required for bacterial host colonization and increased production of virulence factors. Bacterial virulence factors, as well as free-heme, activates the inflammasome, contributing to neutrophil inflammation, cytokine elevation, and pathogenesis processes. This figure was adapted from "SARS-CoV-2: What We Know About Its Effects on Respiration", by BioRender. com (2020). Retrieved from https://app.biorender.com/biorender-templates.
Data in the literature showed an association between ventilatorassociated pneumonia and biofilm production due to microbial interaction on endotracheal tubes [31,32]. Moreover, in clinical isolates with persistent infections, P. aeruginosa, S. aureus, and C. albicans predominated as the most biofilm-producers [33-35]. Furthermore, when in biofilm, bacterial species showed more resistance to immune response, evading neutrophil-mediated phagocytosis [36] and enriching macrophage secretory products oxygen-limiting conditions [37,38].
During biofilm production, metabolites of the TCA cycle have shown to be up-regulated [39]. Decreasing in biofilm accumulation was related to an increase in antibiotics susceptibility, as consequence of disruption in enoyl-CoA hydratase activity [40]. Enoyl-CoA hydratase catalyzes the first step of fatty acid oxidation, and it has been reported that Escherichia coli employs this pathway Hemolysin is a critical virulence factor of E. coli, S. aureus, and Enterococcus faecalis
Besides enoyl-CoA hydratase enzyme, we highlighted the identification of dihydrolipoamide dehydrogenase, phosphoglucosamine mutase, UDP-N-acetylglucosamine, sortase, cyto- or haemo-lysin, and iron/heme acquisition proteins, as other virulence factors that could be involved in the clinical worsening of COVID-19 patients. Dihydrolipoamide dehydrogenase plays an important role in pneumococcal infection by Streptococcus pneumoniae, being necessary for survival and capsular polysaccharide production of pneumococci within-host [42]. Phosphoglucosamine mutase acts on the first step in the cell wall biosynthetic pathway leading to the formation of essential peptidoglycan precursor UDP-N-acetylglucosamine, which is involved in a variety of cellular functions, such as cell adhesion or signal recognition [43]. In Staphylococcus aureus, phosphoglucosamine mutase has been a potential candidate for inhibition drug resistance [43]. Additionally, a virulence role of sortase has been shown to mediate adhesion to host tissues, host cell entry, evasion, and suppression of immune response [44].
RTX agglutinin and streptolysins are hemolysins that disrupt erythrocytes, leading to impaired phagocytic clearance and epithelial cell cytotoxicity [45,46]. Hemolysin is a critical virulence factor of E. coli, S. aureus, and Enterococcus faecalis [47,48]. Hemolysin, polysaccharide capsule proteins, and adhesins reduce the Streptococcus pneumoniae entrapment in the pulmonary mucus, permitting its adherence, leading to lung injury, and induction of interleukin (IL)–8 [49,50]. In Acinetobacter spp., Harding and authors demonstrated that Type I Secretion System is required for RTX-toxin exporting, and proteins of this system participate in biofilm formation, with attenuated virulence phenotype observed in T1SS Acinetobacter mutants [51].
Third, mechanisms for iron acquisition are linked to bacterial growth, virulence, and host immunological and clinical disorders. It was demonstrated that free-heme and iron derived from erythrocyte disruption induced by pathogens is required for their invasion and successful colonization in the host, having an essential role during infection [52]. Pseudomonas aeruginosa uses the secreted hemophore HasAp (T1SS) to capture extracellular heme, taken up by an outer membrane TonB-dependent receptor [53]. In high iron levels, aggregation, adhesion, and biofilm production are stimulated [54]. Under iron-rich and -chelated conditions, induction of iron-proteins such as the SdhA family was evidenced in A. baumannii [55]. Moreover, the acquisition of heme-iron bound to erythrocyte hemoglobin is the preferred source of iron during S. aureus infection, promoting Staphylococcal cellular adherence to host cells and resistance to neutrophil killing [56-58]. Furthermore, Gupta et al. demonstrated an increase of hemolysin, toxins, and biofilm formation in a positive Fe-dependent regulation during pneumonia and bacteremia occasioned by Streptococcus pneumoniae [59].
In serum presence, heme and microbial effectors in synergy trigger cytokine production, promoting processing of caspase-1, inflammasome activation, and IL-1β secretion [60] (Figure 6). Besides that, bacterial pathogens had evolved other strategies for inflammasome activation, resulting in secretion of IL-1β and IL-18 and apoptotic and pyroptotic cell death [61].
Furthermore, iron or heme-free has been associated with important clinical conditions of patients with severe pulmonary diseases. In Chronic Obstructive Pulmonary Disease (COPD) and ARDS, heme uptake was involved in vascular and coagulation disorders, lung injury, increase of cell-free Hemoglobin (Hb), and HO-1 expression [62-64]. Additionally, ACE-like activity of Hb has been demonstrated, with vasoconstriction induced by ferrousand ferryl-Hb [65]. In the blood of severe COVID-19 patients, heme and Heme Oxygenase (HO)-1 expression are increased [66], and an additional role of hemoglobinopathy, hypoxia, and hyperferritinemia has been proposed in worsening COVID-19 infection [67]. A blood transcriptome-based analysis of coronavirusinfected patients aiming to infer SARS-CoV-2 gene signatures to the whole organism showed that, outside of the inflammatory response, genes associated with iron/heme transport had increased expression [68]. Our data are in accordance with Sadanandam et al. [68] and highlighted for investigation of the presence of bacterial pathogens in COVID-19 patients admitted to ICU, which together with broad-spectrum antibiotic use is a trigger for the disease that should not be neglected.
Conclusion
We have shown respiratory pathogens as a critical component of pulmonary microbiota of patients admitted to ICU with COVID-19. The extensive diversity of virulence factors found was linked to a possible worsening of the immune response. Functional findings reported providing a better understanding of how bacterial pathogens can aggravate the clinical condition of patients admitted to intensive care units. Our data allow intriguing insights that can assist how bacterial interactions can act synergistically, favoring a cyclic hyperactivated inflammatory response, contributing to COVID-19 severity.
Author Contributions
Conceptualization and Methodology, A.T.R.V., R.S.A. C.C.C., F.M.C., and L.N.L; Investigation, F.M.C., L.N.L., L.P.C.; Writing –Original Draft, F.M.C. and L.N.L.; Writing –Review and Editing, C.C.C., A.T.R.V and R.S.A.; Funding Acquisition, A.T.R.V.; Resources, R.G.M., A.G., A.P.C.G., T.F., V.A.A.Z., R.A., T.B.A., J.S.L., S.P.C.B, M.M.T., R.P.S.; Supervision, A.T.R.V.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of Marcílio Dias Navy Hospital and Eduardo Menezes Hospital (protocol numbers 32382820.3.0000.5256 and 31462820.3.0000.5149).
Data Availability
NGS data generated in our study is publicly available in SRA-NCBI (www.ncbi.nlm.nih.gov/sra), Bioproject accession PRJNA683652.
Funding
This work was developed under the frameworks of Corona-ômica-RJ (FAPERJ=E-26/210.179/2020) and Rede Corona-ômica BR MCTI/ FINEP (FINEP=01.20.0029.000462/20, CNPq=404096/2020-4). A.T.R.V. is supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPq (303170/2017-4) and FAPERJ (E -26/202.826/2018). R.S.A. is supported by CNPQ 312688/2017- 2, 439119/2018-9; FAPERJ 202.922/2018. C.C.C is supported by FAPERJ (E-26/202.791/2019) and S.P.C.B is supported by FAPERJ (E-26/010.000168/20).
REFERENCES
- Bourgonje AR, Abdulle AE, Timens W, Hillebrands JL, Navis GJ, Gordijn SJ, et al. Angiotensin‐converting enzyme 2 (ACE2), SARS‐CoV‐2 and the pathophysiology of coronavirus disease 2019 (COVID‐19). J Pathol. 2020;251(3):228-248.
- Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31(4):S195-S199.
- Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017;39(5):529-539.
- Arachchillage DR, Laffan M. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. Journal of thrombosis and haemostasis. 2020;18(5):1233-1234.
- West JB. Regional differences in the lung. Chest. 1978;74(4):426-437.
- Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet. 2014;384(9944):691-702.
- Balamayooran G, Batra S, Fessler MB, Happel KI, Jeyaseelan S. Mechanisms of neutrophil accumulation in the lungs against bacteria. Am J Respir Cell Mol Biol. 2010;43(1):5-16.
- Klein EY, Monteforte B, Gupta A, Jiang W, May L, Hsieh YH, et al. The frequency of influenza and bacterial coinfection: a systematic review and meta‐analysis. Influenza Other Respir Viruses. 2016;10(5):394-403.
- Zahariadis G, Gooley TA, Ryall P, Hutchinson C, Latchford MI, Fearon MA, et al. Risk of ruling out severe acute respiratory syndrome by ruling in another diagnosis: variable incidence of atypical bacteria coinfection based on diagnostic assays. Can Respir J. 2006;13(1):17-22.
- Cendrero JA, Solé-Violán J, Benitez AB, Catalán JN, Fernández JA, Santana PS, et al. Role of different routes of tracheal colonization in the development of pneumonia in patients receiving mechanical ventilation. Chest. 1999;116(2):462-470.
- Gil-Perotin S, Ramirez P, Marti V, Sahuquillo JM, Gonzalez E, Calleja I, et al. Implications of endotracheal tube biofilm in ventilator-associated pneumonia response: a state of concept. Crit Care. 2012;16(3):1-9.
- Vandecandelaere I, Matthijs N, Van Nieuwerburgh F, Deforce D, Vosters P, De Bus L, et al. Assessment of microbial diversity in biofilms recovered from endotracheal tubes using culture dependent and independent approaches. PloS one. 2012;7(6):e38401.
- Vaillancourt M, Jorth P. The unrecognized threat of secondary bacterial infections with COVID-19. Mbio. 2020;11(4).
- Bengoechea JA, Bamford CG. SARS‐CoV‐2, bacterial co‐infections, and AMR: the deadly trio in COVID‐19?. EMBO Mol Med. 2020;12(7):e12560.
- Manohar P, Loh B, Nachimuthu R, Hua X, Welburn SC, Leptihn S. Secondary bacterial infections in patients with viral pneumonia. . Front Med. 2020;7:420.
- Zhu X, Ge Y, Wu T, Zhao K, Chen Y, Wu B, et al. Co-infection with respiratory pathogens among COVID-2019 cases. Virus Res. 2020;285:198005.
- Langford BJ, So M, Raybardhan S, Leung V, Westwood D, MacFadden DR, et al. Bacterial co-infection and secondary infection in patients with COVID-19: a living rapid review and meta-analysis. Clin Microbiol Infect. 2020.
- Rawson TM, Moore LS, Zhu N, Ranganathan N, Skolimowska K, Gilchrist M, et al. Bacterial and fungal coinfection in individuals with coronavirus: a rapid review to support COVID-19 antimicrobial prescribing. Clin Infect Dis. 2020;71(9):2459-68.
- Ranzani OT, Bastos LS, Gelli JG, Marchesi JF, Baião F, Hamacher S, et al. Characterisation of the first 250 000 hospital admissions for COVID-19 in Brazil: a retrospective analysis of nationwide data. The Lancet Resp. Med. 2021;9(4):407-418.
- Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet. 2019;20(6):341-355.
- Chakraborty S. Metagenome of SARS-Cov2 from a patient in Brazil shows a wide range of bacterial species-Lautropia, Prevotella, Haemophilus-overshadowing viral reads, which does not even add up to a full genome, explaining false negatives.2020.
- Meyer F, Paarmann D. D'S, ouza M, Olson R, Glass EM, Kubal M, et al. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinforma. 2008;9:386.
- McMurdie PJ. 8c Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of mi-crobiome census data. PLoS One. 2013;8:e61217.
- Shetty SA, Lahti L. Microbiome data science. J Biosci. 2019;44(5):1-6.
- Wei T, Simko V. R package "corrplot": Visualization of a Correlation Matrix. 2017.
- Wickham H, Chang W, Henry L, Pedersen TL, Takahashi K, Wilke C, et al. Springer-Verlag. New York. 2016.
- Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Beck JM, Huffnagle GB, Curtis JL. Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann Am Thorac Soc. 2015;12(6):821-830.
- Harding CM, Hennon SW, Feldman MF. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat Rev Microbiol. 2018;16(2):91.
- Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008;6(4):288-301.
- Bien J, Sokolova O, Bozko P. Characterization of virulence factors of Staphylococcus aureus: novel function of known virulence factors that are implicated in activation of airway epithelial proinflammatory response. J Pathog. 2011.
- Adair CG, Gorman SP, Feron BM, Byers LM, Jones DS, Goldsmith CE, et al. Implications of endotracheal tube biofilm for ventilator-associated pneumonia. Intensive Care Med. 1999;25(10):1072-1076.
- Wolcott R, Costerton JW, Raoult D, Cutler SJ. The polymicrobial nature of biofilm infection. . Clin Microbiol Infect. 2013;19(2):107-112.
- Boisvert AA, Cheng MP, Sheppard DC, Nguyen D. Microbial biofilms in pulmonary and critical care diseases. Ann Am Thorac Soc. 2016;13(9):1615-1623.
- Sanchez CJ, Mende K, Beckius ML, Akers KS, Romano DR, Wenke JC, Murray CK. Biofilm formation by clinical isolates and the implications in chronic infections. BMC Infect Dis. 2013;13(1):1-2.
- Marzetti S, Carranza C, Roncallo M, Escobar GI, Lucero NE. Recent trends in human Brucella canis infection. Comp Immunol Microbiol Infect Dis. 2013;36(1):55-61.
- Lu H, Que Y, Wu X, Guan T, Guo H. Metabolomics deciphered metabolic reprogramming required for biofilm formation. . Sci Rep. 2019;9(1):1-7.
- Cameron LC, Bonis B, Phan CQ, Kent LA, Lee AK, Hunter RC. A putative enoyl-CoA hydratase contributes to biofilm formation and the antibiotic tolerance of Achromobacter xylosoxidans. NPJ biofilms and microbiomes. 2019;5(1):1-7.
- Hirschfeld J. Dynamic interactions of neutrophils and biofilms. J Oral Microbiol. 2014;6(1):26102.
- Borriello G, Werner E, Roe F, Kim AM, Ehrlich GD, Stewart PS. Oxygen limitation contributes to antibiotic tolerance of Pseudomonas aeruginosa in biofilms. Antimicrob Agents Chemother. 2004 ;48(7):2659-2664.
- Hanke ML, Kielian T. Deciphering mechanisms of staphylococcal biofilm evasion of host immunity. Front Cell Infect Microbiol. 2012;2:62.
- Campbell JW, Morgan‐Kiss RM, E. Cronan Jr J. A new Escherichia coli metabolic competency: growth on fatty acids by a novel anaerobic β‐oxidation pathway. Mol Microbiol. 2003;47(3):793-805.
- Smith AW, Roche H, Trombe MC, Briles DE, Håkansson A. Characterization of the dihydrolipoamide dehydrogenase from Streptococcus pneumoniae and its role in pneumococcal infection. Mol Microbiol. 2002;44(2):431-448.
- Hou Y, Mayhood T, Sheth P, Tan CM, Labroli M, Su J, et al. NMR binding and functional assays for detecting inhibitors of S. aureus MnaA. J Biomol Screen. 2016;21(6):579-589.
- Spirig T, Weiner EM, Clubb RT. Sortase enzymes in Gram‐positive bacteria. Mol Microbiol. 2011 ;82(5):1044-1059.
- Chu F, Kearns DB, Branda SS, Kolter R, Losick R. Targets of the master regulator of biofilm formation in Bacillus subtilis. Mol Microbiol. 2006;59(4):1216-1228.
- Liu PV, Shokrani FA. Biological activities of pyochelins: iron-chelating agents of Pseudomonas aeruginosa. Infec Immu. 1978;22(3):878-890.
- Wiseman GM. The hemolysins of Staphylococcus aureus. Bacteriol Rev. 1975;39(4):317.
- Schindel C, Zitzer A, Schulte B, Gerhards A, Stanley P, Hughes C, et al. Interaction of Escherichia coli hemolysin with biological membranes: A study using cysteine scanning mutagenesis. Eur J Biochem. 2001;268(3):800-808.
- Ike YA, Hashimoto HA, Clewell DB. High incidence of hemolysin production by Enterococcus (Streptococcus) faecalis strains associated with human parenteral infections. J Clin Microbiol. 1987 ;25(8):1524-1528.
- Doran KS, Chang JC, Benoit VM, Eckmann L, Nizet V. Group B streptococcal β-hemolysin/cytolysin promotes invasion of human lung epithelial cells and the release of interleukin-8. . J Infect Dis. 2002;185(2):196-203.
- Harding CM, Pulido MR, Di Venanzio G, Kinsella RL, Webb AI, Scott NE, et al. Pathogenic Acinetobacter species have a functional type I secretion system and contact-dependent inhibition systems. J Biol Chem. 2017;292(22):9075-9087.
- Richard KL, Kelley BR, Johnson JG. Heme uptake and utilization by gram-negative bacterial pathogens. Front Cell Infect Microbiol. 2019;9:81.
- Dent AT, Mouriño S, Huang W, Wilks A. Post-transcriptional regulation of the Pseudomonas aeruginosa heme assimilation system (Has) fine-tunes extracellular heme sensing. J Biol Chem. 2019;294(8):2771-5555.
- Kang D, Kirienko NV. Interdependence between iron acquisition and biofilm formation in Pseudomonas aeruginosa. J Microbiol. 2018;56(7):449-457.
- Nwugo CC, Gaddy JA, Zimbler DL, Actis LA. Deciphering the iron response in Acinetobacter baumannii: a proteomics approach. J Proteomics. 2011;74(1):44-58.
- Weinberg ED. Iron and infection. Microbiol Rev. 1978;42(1):45.
- Skaar EP. Staphylococcus aureus Iron-Source Preference of. science. 2004;1099930(1626):305.
- Ledala N, Zhang B, Seravalli J, Powers R, Somerville GA. Influence of iron and aeration on Staphylococcus aureus growth, metabolism, and transcription. J Bacteriol. 2014;196(12):2178-2189.
- Gupta R, Shah P, Swiatlo E. Differential gene expression in Streptococcus pneumoniae in response to various iron sources. Microb Pathog. 2009;47(2):101-109.
- Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, et al. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci USA. 2014;111(39):E4110-E4118.
- Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov. 2020;6(1):1-22.
- M Raval C, J Lee P. Heme oxygenase-1 in lung disease. Current drug targets. 2010;11(12):1532-1540.
- Mumby S, Upton RL, Chen Y, Stanford SJ, Quinlan GJ, Nicholson AG, et al. Lung heme oxygenase-1 is elevated in acute respiratory distress syndrome. Crit Care Med. 2004;32(5):1130-1135.
- Shaver CM, Upchurch CP, Janz DR, Grove BS, Putz ND, Wickersham NE, et al. Cell-free hemoglobin: a novel mediator of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2016;310(6):L532-L541.
- Simoni J, Simoni G, Moeller JF, Tsikouris JP, Wesson DE. Evaluation of angiotensin converting enzyme (ACE)-like activity of acellular hemoglobin. Artif Cells Blood Substit Immobil Biotechnol. 2007;35(2):191-210.
- Su WL, Lin CP, Hang HC, Wu PS, Cheng CF, Chao YC. Desaturation and heme elevation during COVID-19 infection: A potential prognostic factor of heme oxygenase-1. J Microbiol Immunol Infect. 2021;54(1):113-116.
- Wagener FA, Pickkers P, Peterson SJ, Immenschuh S, Abraham NG. Targeting the heme-heme oxygenase system to prevent severe complications following COVID-19 infections. Antioxidants. 2020;9(6):540.
- Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Death Discov. 2020;17(6):613-620.
Citation: de Carvalho FM, Lemos LN, Ciapina LP, Moreira RG, Gerber A, Guimaraes APC, et al. (2021) Insights of How Lung Microbiome can Contribute to COVID-19 Severity in Intensive Care Unit Patients. J Vaccines Vaccin. 12:450.
Copyright: © 2021 de Carvalho FM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.