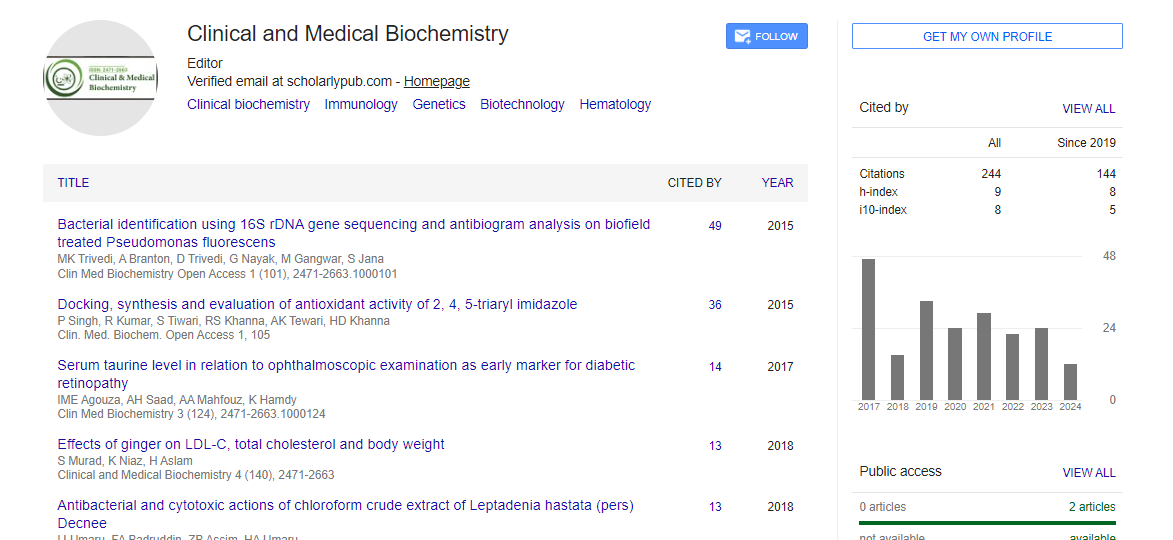

Indexed In
- RefSeek
- Directory of Research Journal Indexing (DRJI)
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Scholarsteer
- Publons
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Perspective - (2024) Volume 10, Issue 2
Hemoglobinopathy: Genetic Differences Noted in the Hemoglobin Perspective
Sarah Ashley*Received: 25-Apr-2024, Manuscript No. CMBO-24-25759; Editor assigned: 29-Apr-2024, Pre QC No. CMBO-24-25759 (PQ); Reviewed: 14-May-2024, QC No. CMBO-24-25759; Revised: 22-May-2024, Manuscript No. CMBO-24-25759 (R); Published: 30-May-2024, DOI: 10.35841/2471-2663.24.10.217
Description
Abnormalities in the structure or synthesis of hemoglobin, the oxygen-carrying protein present in red blood cells, identify a wide range of hereditary illnesses known as hemoglobinopathies. Because of these diseases' high prevalence, high morbidity, and profound effects on afflicted people and their families, including sickle cell disease and other types of thalassemia, they pose serious problems for healthcare systems around the globe. Studying the hemoglobinopathies' genetic, clinical, and therapeutic aspects helps us get to know the complexity of these disorders and the continuous efforts to treat their various presentations. Genetic mutations that impact the genes that encode the hemoglobin components are the source of hemoglobinopathies. A broad range of clinical symptoms can be caused by these mutations, which can change the hemoglobin's synthesis or structure. For example, a particular point mutation in the β-globin gene causes sickle cell disease by producing hemoglobin S, an abnormal form of hemoglobin. Red blood cells with this hemoglobin type take on the typical sickle shape when low oxygen tension causes it to polymerize. The disease's clinical symptoms become more severe by this unusual shape, which reduces blood flow and oxygen delivery.
Defects in the production of globin chains cause thalassemia, another category of hemoglobinopathies. There are two types of thalassemia: α-thalassemia and β-thalassemia, or their variations, depending on the particular mutation and how it affects the formation of the globin chain. Reduced α-globin chain synthesis results in an excess of β-globin chains and the production of unstable hemoglobin tetramers in α-thalassemia. On the other hand, decreased β-globin chain synthesis causes an excess of α- globin chains to precipitate within red blood cells, causing hemolysis and anemia in β-thalassemia. Hemoglobinopathies exhibit clinically a wide range of symptoms and implications that impact several organ systems. Premature red blood cell breakdown leads to chronic hemolytic anemia, which is a characteristic of numerous hemoglobinopathies. Patients' blood's diminished ability to carry oxygen may cause them to feel lethargic, pale, and jaundiced. Vaso-occlusive episodes can also cause tissue ischemia and organ damage, with possible long-term effects like stroke and pulmonary hypertension. One example of this is suffering in sickle cell disease.
Hemoglobinopathies need to be diagnosed and treated with multiple approaches which involve specialist imaging inquiries, laboratory testing, and clinical evaluation. Programs for newborn screening are essential for early detection, which enables quick action that decreases the effects of these illnesses on those who are affected. Hemoglobin electrophoresis, genetic testing, and clinical evaluation are frequently used in the diagnosis process to define the particular hemoglobinopathy and provide therapy choices. The objectives of hemoglobinopathy treatment plans are to reduce symptoms, stop impacts, and enhance the lives of those who are impacted. Transfusions of blood are frequently used to treat anemia and relieve symptoms, especially in those with severe thalassemia. To avoid organ damage, however, prolonged transfusion therapy poses a risk of iron overload and transfusion-related problems, resulting in the need for close monitoring and supplemental treatments such as iron chelation.
Recent developments in gene therapy and molecular biology have given hemoglobinopathy patients fresh hope. The basic mutations causing these ailments may be corrected by gene editing technologies like CRISPR-Cas9, providing possibilities for potentially curative medicines that target the fundamental cause of disease. Positive results from clinical trials exploring gene therapy techniques for β-thalassemia and sickle cell disease have indicated that these novel therapies are both practical and efficient. In addition, the development of precision medicine has completely changed the way hemoglobinopathies are managed by developing therapy plans based on each patient's unique genetic and clinical characteristics. Treatments that target particular molecular pathways, like hydroxyurea for sickle cell disease, have been suggested to be effective in symptom relief and in the prevention of consequences. In a similar vein, new drugs that alter globin chain synthesis show promise in treating thalassemia patients by balancing out the α- and β-globin chains and improving anemia.
Conclusion
It should be noted that hemoglobinopathies are a difficult and complicated category of hereditary illnesses that have a big impact on patients, families, and medical professionals. By investigating the genetic, clinical, and therapeutic aspects of these disorders, we can discover important things about their fundamental causes and the continuing attempts to create efficient treatments. To address the various hemoglobinopathy symptoms and enhance outcomes for those affected globally, a multidisciplinary relationship and creative thinking will be necessary as research advances.
Citation: Ashley S (2024) Hemoglobinopathy: Genetic Differences Noted in the Hemoglobin Perspective. Clin Med Bio Chem. 10:217.
Copyright: © 2024 Ashley S. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.