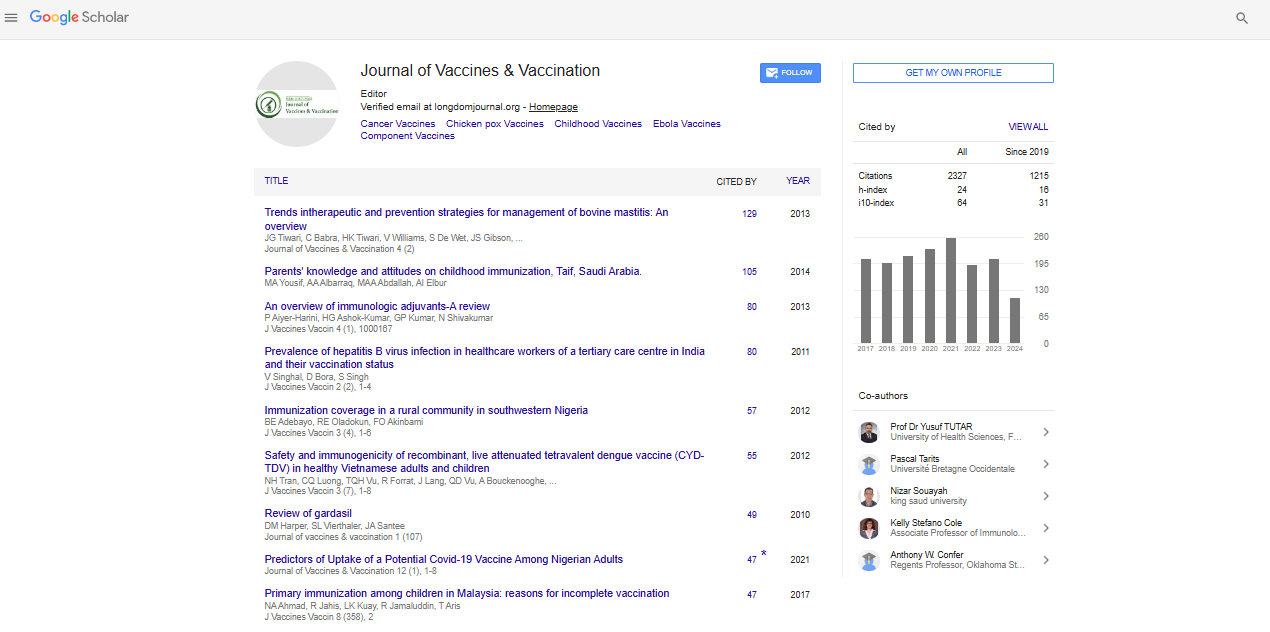

Indexed In
- Academic Journals Database
- Open J Gate
- Genamics JournalSeek
- JournalTOCs
- China National Knowledge Infrastructure (CNKI)
- Scimago
- Ulrich's Periodicals Directory
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- MIAR
- University Grants Commission
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2014) Volume 5, Issue 3
Genomic CpG Enrichment of Oncolytic Parvoviruses as a Potent Anticancer Vaccination Strategy for the Treatment of Pancreatic Adenocarcinoma
Abstract
Objective: For quite a long time oncolytic viruses (OVs) have been regarded merely as specific tumor cell killers, while disregarding the fact that all oncolytic activities take place in the context of a functional immune system. Oncolytic parvoviruses (PV) represent non-pathogenic, naturally oncolytic (non-modified), animal (rodent) viruses with a tropism that extends to a number of transformed human cells. Our recent work using various animal models substantiates the contention that H-1PV acts as both an oncolytic agent and an adjuvant, by direct cytoreduction in the tumor and bystander antitumor immunity. ImmunostimulatoryCpG motifs were incorporated into the singlestranded DNA genome of H-1PV and our current objective was to test whether the CpG-armed virus was in possession of an enhanced adjuvant capacity. Methods: The immunogenic potential of the CpG-enriched parvoviral derivative (JabCG) was tested in in vitro infection of human PBMCs or coculture of DCs and T-cells. In vivo tumor xenografts were raised in NOD.SCID mice that were later reconstituted with an autologous DCs and T-cells mix primed with an infected or chemovirotherapy (gemcitabine and H-1PV)-treated pancreatic cancer line vaccine. The therapeutic activity of the native and modified viruses was evaluated upon systemic application in pancreatic cancer-bearing immunocompetent Lewis rats. Results: Compared with wt H-1PV, JabCG displayed enhanced immunotherapeutic capacity to activate human immune cells ex vivo (PBMCs or DCs and T-cells isolated from pancreatic cancer patients) with a striking increase in the capacity of the latter cells for suppressing autologous tumorxenografts in NOD.SCID mice. Furthermore, intravenous application of JabCG in immunocompetent rats caused early NK and T-cell infiltration into tumors, elevated IFNγ levels in serum and spleens, and notably prolonged survival, as compared to control-treated animals. Conclusion: Taken together, data indicate that CpG-enrichment of OVs represents a potent strategy to enhance their immunotherapeutic properties.
Keywords: Parvovirus H-1PV, CpG motifs, Pancreatic cancer, Immune respons, Ssystemic application, Immunomodulation
Abbreviations
OV: Oncolytic Virus; PV: ParvoVirus; wt H-1PV: wild type H-1 ParvoVirus; JabCG: CpG enriched H-1PV; PDAC: Pancreatic AdenoCarcinoma; PBMCs: Peripheral Blood Monocytes; DC: Dendritic Cell; pfu: plaque forming units; dpi: days post infection; ICD: Immunogenic Cell Death; CVT: Chemovirotherapy; αH-1PV: anti H-1PV neutralizing antibodies; IP-10: interferon gamma-induced protein 10; 2,5-OAS: 2'-5'-Oligoadenylate Synthetase; ISG-15: Interferon-Induced Gene 15; IFITM1: Interferon-Induced Transmembrane Protein 1
Introduction
Oncolytic viruses (OVs) are a novel class of anticancer agents that attract increasing clinical attention. Currently, more than 20 viruses have been shown to possess oncotropic and oncolytic features. In the last years, compelling evidence accumulated that the small autonomous parvoviruses (PVs) can achieve complete cure of various tumors in animal models, and kill tumor cells that resist conventional anticancer treatments [1]. wt H-1PV is currently subjected to a phase I/IIa clinical trial, in which the virus is administered to patients with recurrent glioblastomamultiforme [2].
The work of our group is focused on the design and production of new PV variants, and their comparison with wt H-1PV for use as components of immunomodulating combinations [3]. As recently demonstrated the adjuvant capacity of oncolytic H-1PV can be increased by incorporating immunostimulatory CpG motifs into the viral single-stranded DNA genome in order to enhance innate immune TLR-9 signalling [4,5]. Virus-mediated delivery of the immune-priming CpG elements to tumor cells leads to their further amplification together with the viral genome. Antitumor activity was evaluated in a protocol where cells were infected with wt H-1PV or CpG-modified (JabCG) viruses, lethally irradiated and administered subcutaneously to serve as an autologous therapeutic vaccine for the treatment of hepatoma lung metastases. In this setup, the virus essentially acts as an ex vivo administered adjuvant to the vaccine. The JabCG virus was found to be more effective than the wt H-1PV in potentiating the vaccine, as evidenced by a reduction of metastatic rates correlating with the induction of markers of cellular immunity and dendritic cells activation.
There is strong evidence to suggest that H-1PV can modulate the immune status of PBMCs. This occurs both in a direct way by inducing the abortive infection of distinct subsets of these cells, and in an indirect way by infecting tumor cells with which the PBMCs are brought into contact [6-9]. Both procedures were found to cause a shift of the PBMC immune balance in favour of a cellular Th1 response [9-11]. Therefore, H-1PV appears to exert immunostimulating activities through both direct immune cell stimulation and immunogenic tumor cell death induction. It should also be stated that besides their role in cancer immune surveillance, PBMCs may serve as carriers for OVs upon systemic virus injection. This is the case of oncolyticreoviruses which were recently reported to be present in the human bloodstream as both free particles in the plasma and viruses bound to peripheral blood monocytes, granulocytes and platelets [12].
Adoptive transfer [9], immunodepletion [10] and rechallenge [13] experiments indicate that PV-mediated oncosuppression involves an immunological component. As recently reported the wt H-1PV-induced suppression of pancreatic cancer (PDAC) in an orthotopic animal model correlates with immune stimulation. This argues in favour for the contribution of above-mentioned immunomodulating effects of H-1PV to tumor eradication. However, it remains to be demonstrated that ex vivo activation through direct or tumor cell-mediated H-1PV infection provides PBMCs with enhanced anti-cancer potency. This essential issue was addressed in the present work, using autologous pancreatic cancer cells and PBMCs newly derived from the same patient to achieve tumor formation and immune reconstitution in NOD, SCID mice, respectively. wt H-1PV and, to a greater extent, the JabCG derivative proved able to stimulate human anti-cancer immune cells ex vivo, as revealed by a stronger suppression of autologous pancreatic tumors in animals reconstituted with virus (vs. mock)-treated PBMCs. These data indicate that H-1PV has an immunostimulating capacity which can be reinforced through appropriate genetic engineering, and plays an important role in virus-mediated tumor suppression. In agreement with this conclusion, JabCG was found to be more efficient than the wild-type virus in inhibiting the development of rat PDAC in an immunocompetent rat model treated through intravenous virus administration.
Materials and Methods
Cells and virus
Human pancreatic carcinoma cell lines Panc-1, and T3M4 were obtained from ATCC (Manassas, VA) and grown in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS). The DSL-6A/C1 cell line derived from a chemically induced pancreatic ductal adenocarcinoma in Lewis rats was grown in DMEM supplemented with 10% FCS. Human NB324K cells used for cytotoxicity protection assays were cultured in MEM medium with 5% FCS. All media were supplemented with penicillin (100 U/ml) and streptomycin (100 µg/ml).
Peripheral blood mononuclear cells (PBMC)
Peripheral blood mononuclear cells (PBMC) were isolated from the heparinized blood of either randomly selected healthy donors or PDAC patients by differential centrifugation over Histopaque (Sigma) and cultured in RPMI with 10% FCS and antibiotics. Buffy coats were obtained from the blood bank of IKTZ Heidelberg. Patient blood was collected approximately 6 months after obtaining the tumor biopsy and prepared either as PBMCs or further processed for the generation of DCs and T-cells.
Dendritic cells
Dendritic cells were generated from peripheral blood samples afterincubation of plastic-adherent cells for 3 days with X-Vivo 20 medium supplemented with GM-CSF and IL-4as described previously [14]. T3M4 cells were either left untreated or treated with Gemzar (1.2 ng/ml) and infected with H-1PV at an MOI of 10 (CVT protocol) or plainly infected (H-1PV or JabCG at an MOI of 10) for comparison of viral adjuvant effects. Mock, infected or CVT treated cells were irradiated with 30Gy. 48hrs post treatment T3M4:DCs (1:10) cocultures were set for 72hrs when lysis of tumor cells and maturation of DCs occur. Afterwards supernatants were collected for cytokine evaluation by qPCR.
T lymphocytes
T lymphocytes were obtained from remaining PBMCs after DC preparation and incubated inT cell medium (RPMI-1640 supplemented with 10% AB Serum, IL-4 60 U/ml and IL-2 100 U/ml) until use. Enrichmentof T cell populations was performed usingmagnetic beads coupled with mAbs (MiniMACS Miltenyi, Bergisch Gladbach, Germany) according to the manufacturer's instructions.
Transwell system
NBK or T3M4 cells were plated in 6 well plates at 2 x 104 cells per well. PBMCs were infected with H-1PV (MOI10) for 48hrs and washed three times with PBS and cultured within the transwell system pore size 0.4 µm (BD Falcon, USA). The co-cultivation took place for 72 hrs and the cell survival rates were assessed using a MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) assay (Sigma, USA).
Virus-neutralizing antibody detection
Serial dilutions of the sera of experimental animals were made in MEM and mixed with an equal volume of H-1PV virus suspension (corresponding to 2 x 104 pfu/well). After incubation for 30 min. at 37°C, the mixture was inoculated onto NB324K cells plated in 96-well plates (2 x 10^3 cells/well). The cell survival rates were assessed after 72h using a MTT assay.
Viruses (H-1PV, JabCG) were produced in NBK cell and purified to GLP grade by Virus Production Unit at DKFZ-Heidelberg.
qPCR and semi-quantitative RT-PCR
For RT-PCR and qPCR total RNA was extracted from DCs and PBMC, mono- or cocultures with T3M4 cells, pancreatic tumors, liver metastatic nodules, spleens and lymph nodes of treated animals and reverse transcribed into cDNA. For qPCR all reagents and equipment for mRNA/cDNA preparation were supplied by Roche Applied Science (Mannheim, Germany). mRNA was prepared by automated isolation using the MagNA pure LC instrument and isolation kit I. All indicated values are normalized to cyclophilin B.The primers used to amplify rat IFNγ cDNAs were 5’-ATCTGGAGGAACTGGCAAAAGGACG-3’-forward, and 5’-CCTTAGGCTAGATTCTGGTGACAGC-3’-reverse, resulting in a product of 290 bp. For TNF 5’-TACTGAACTTCGGGGTGATCGGTCC-3’-forward and 5’-CAGCCTTGTCCCTTGAAGAGAACC-3’ –reverse resulting in a product of 295 bp. Primer sequences for detection of H-1PV in QRT-PCR and RT-PCR, and ß-actin expression in RT-PCR have been previously published [18].
ELISA. Measurement of rat TNF, IL-2 and IFNγ as well as human TNF, IL-1,2 and IFN-,-,-γ release was done using commercially available ELISA kits from eBioscience (Frankfurt, Germany) as described by manufacturer.
Flowcytometry
Tumors were pressed against a mesh to obtain single cell suspensions and were adjusted to a concentration of 5 x 106/ml in PBS. At the time-points indicated in Figures, single cell suspensions from tumors or cultured human PBMC, were harvested and washed once in ice-cold PBS. For discrimination of monocyte and lymphocyte subpopulations during flow cytometry analysis, fluorochrome-conjugated MAbs against the following surface determinants were used: anti-rat CD3, CD8, CD4, CD25, anti-NK; anti-human CD3, CD8, CD4, CD69, CD14, CD80, CD86, CD19, CD20, HLA-DR, CD11c, CD45R (Immunotools, Friesoythe, Germany). The cells were first blocked either with 10% FCS for rat splenocytes, or FcR blocking reagent (MiltenylBiotec GmbH, Bergisch Gladbach, Germany) for human PBMCs. Cells were then incubated with monoclonal antibodies or appropriate FITC-, PE- or APC-conjugated isotype controls for 30 min on ice, washed twice in ice-cold PBS and used for flow cytometry acquisition. Typically, list mode data for 30,000 events for PBMCs or 20,000 events for rat single cell suspension in a "live-gate" mode were acquired. Samples were analyzed in a FACS Calibur flow cytometer (Becton Dickinson) by using Cellquest software.
Animal studies
The orthotopic rat model using DSL-6A/C1 cells has been previously described [16]. A cell suspension was prepared in phosphate-buffered saline (PBS) out of subcutaneous tumors preformed by implantation of DSL-6A/C1 cells and injected intrapancreatically to Lewis rats. For the induction of liver cancer in parallel intrahepatic inoculation of 1 x 106 cells in 100 µl per animal was inoculated. For xenotransplantation experiments patients’ tumour material was expanded onto six-week-old C.B.-17/IcrHanHsd-Prkdc-scid mice as described previously [19,20]. Operational tumour material was obtained during explorative laparotomies from patients of the University Clinic Heidelberg, and pathologically confirmed as pancreatic ductal carcinoma (Ethic Votum Nr. 301/2001). Second generation mice bearing xenotransplant tumour of 10-20 mm3 in size (about 1 month after implantation) were inoculated with suspension of DCs and T-cells prepared as described in M&M above. Cells were mixed prior to injection at a ratio of 1:6 (5x105 DC + 3x106 T-cells per animal) and inoculated peritumorally. Tumor development was monitored by measuring tumor size two to three times a week and the tumor volume was calculated for an ellipsoid using the formula V=p/6 × length × width × height (mm3).
Animal experiments were performed according to European Community directives for animal care (No. 86/609/EEC, November 24, 1986).
Statistical methods
In vitro data represent mean values with standard deviations (or standard error) from at least three independent experiments. The statistical significance of differences was assessed by a one tail Student’s unpaired t-test.
Results
JabCG is a more potent inducer of human PBMCs activation than wt H-1PV. It was previously shown that parvovirus infection of PBMCs or of tumor cells cocultured with PBMCs results in the activation of the latter [9]. As a first step in the present study, the activating effects of JabCG and wt H-1PV were compared by infecting healthy donor PBMC mono- or coclutures with the pancreatic cell lines T3M4 or Panc1. Two hallmarks of cellular immune responses, IFNγ and IL-2, were induced to a significantly greater extent by JabCG compared to wt H-1PV, especially in PBMC monocultures (Figure 1). The CpG-armed virus was also stronger than the parental virus in causing the release of some (TNFα in cocultures and type I IFNs in monocultures) but not other (IL-1) inflammatory cytokines (Figure 1). The mRNA levels of interferon-inducible genes (IP-10, ISG15, 2,5-OAS, IFITM1) was measured by qPCR and found to be markedly upregulated by wt H-1PV and even more by JabCG (Table 1).
| CELLS | VIRUS | INTERFERON-INDUCED GENES | |||
|---|---|---|---|---|---|
| IP-10 | ISG15 | OAS | IFITM1 | ||
| PBMC alone | MOCK | 453 | 1218 | 376 | 10417 |
| H-1PV | 11688 | 12483* | 1493* | 12159* | |
| JabCG | 18279* | 13508*,# | 1546*,# | 31657*,# | |
| PBMC+ T3M4 | MOCK | 400 | 17400 | 3034 | 5731 |
| H-1PV | 3324 | 19518 | 3754 | 8660* | |
| JabCG | 27164 | 22114 | 2523 | 34486 | |
Table 1: Cytokine alterations in human PBMCs under mono- or coculture conditions with different virus infections PBMC from healthy donors were plated at 1x106 cells/well alone or in co-culture with PDAC cell lines T3M4 and Panc at 10:1 ratio. Cells were left untreated or were infected with H-1PV or JabCG. Cells were collected at 24h pi and processed for qRT-PCR. Data are represented as cbp normalized number of copies. * - p ≤ 0.05 compared to MOCK, # - p ≤ 0.05 compared to H-1PV.

Figure 1: Infection with JabCG represents a stronger enhancer of cytokine release from PBMCs compared to H-1PV.
The expression of co-stimulatory molecules and activation markers on the surface of immune cells was also investigated by flow cytometry. Infection of PBMC monocultures with both viruses resulted in a clear increase in the percentages of T-helper cells (CD4+) and CTLs (CD8+) positive for the homing receptor CD62L (Figure 2). In addition, infection with wt H-1PV, and more particularly the armed JabCG derivative, correlated with enhanced expression of the co-stimulatory molecule CD86 on all antigen-presenting cell subsets, namely B-cells (CD20+), DCs (CD11c+) and macrophages (CD14+) (Figure 2). CD86 and the T-lymphocyte (CD3) late activation marker CD69 were similarly induced upon infection of PBMC cocultures with T3M4 or Panc1 pancreatic cancer cell lines (Figure 2). Some upregulation of the MHCII surface receptors HLA-DR was also detected after infection of PBMC mono- or cocultures, albeit at a low level (Figure 2).

Figure 2: Expression of activation markers upon H-1PV and JabCG infection of PBMCs.
PBMC from healthy donors were plated at 1x106 cells/well alone or in co-culture with PDAC cell lines T3M4 and Panc at 10:1 ratio. Cells were left untreated or were infected with H-1PV or JabCG at MOI10 or MOI50. Supernatants were collected at 24h or 72h pi, and level of IFNγ(A) (healthy donors, n=20), IL-2 (B) (n=15), TNFα(C) (n=16), IL-1 (D) (n=7), IFNα (E) (n=5), IFNß (F) (n=5) was measured by ELISA.
In summary, these in vitro data confirmed the immunomodulating activity of H-1PV and showed that the JabCG virus provides a more potent danger signal for inducing this stimulation in human immune cells, compared to the parental wt H-1PV.
Parvovirus-infected PBMCs exert a bystander killing effect. We and others have previously shown that H-1PV infection of human PBMCs is abortive and does not result in the production of progeny particles [9]. Yet, PBMCs bind parvoviruses and may contribute to the delivery of input particles into target tumors. This issue is important given that one currently tested route of wt H-1PV administration to cancer patients is through i.v. injection [2]. This prompted the evaluation whether infected human PBMCs can release cell-associated input virions in the absence of progeny virus production. PBMCs stimulated or not with IL-2 were either mock-treated or infected with wt H-1PV or JabCG, and further cultured in transwell plates preseeded with parvovirus-sensitive indicator cells, namely transformed (NBK) or cancer (T3M4, Panc1) ones. Under these conditions, the indicator cells were not in direct contact with the PBMCs but were exposed to parvoviral particles and/or cytokines released from these cells. Parvovirus infection of PBMCs resulted in a marked induction of indicator cell death (Figure 3). It is noteworthy that JabCG-infected PBMCs were more toxic for target cells than H-1PV-infected PBMCs, although both viruses are indistinguishable as regards their direct cytotoxic activity [4]. This suggests that besides released virions, cytokines secreted by infected PBMCs were likely to contribute to the killing of indicator cells. This is in agreement with the above-mentioned greater capacity of JabCG for cytokine induction in PBMCs, as compared with wt H-1PV. Altogether, these results raise the hope that upon systemic application of wt H-1PV in humans, white blood cells will participate in virus transport and delivery, together with toxic cytokines, to target tumors.

Figure 3: PBMCs can serve as a carrier system for parvovirus H-1PV and JabCG.
PBMC from healthy donors were plated at 1x106 cells/well alone or in co-culture with PDAC cell lines T3M4 and Panc1 at a 10:1 ratio. Cells were left untreated or were infected with H-1PV or JabCG at MOI10. Cells were collected after 48h pi, labeled with corresponding antibodies and acquired by FACS. (A), (B) – PBMCs monoculture, (C) – co-culture PBMCs and Panc1, (D) – co-culture PBMCs and T3M4. * - p = 0.05 compared to MOCK, # - p = 0.05 compared to H-1PV.
Parvovirus JabCG can act as an improved cancer vaccine adjuvant in chemovirotherapy-enhanced ex vivo priming of human PBMCs for PDAC suppression. In a next step the anticancer protection of the above-mentioned PV-activated PBMC cultures was tested under in vivo conditions. To this end, the wt H-1PV virus and JabCG derivative were compared for their adjuvant properties by measuring the capacity of human PBMCs exposed ex vivo to infected human PDAC cells, for suppressing pancreatic cancer in an animal model. This model involved NOD.SCID mice that were first implanted with human PDAC xenografts newly derived from patients. Mice were subsequently reconstituted with dendritic and T-cells isolated from ex vivo activated PBMCs originating from the same patient as the tumorxenograft. The dendritic and T-cell mixture used for the immune reconstitution was thus preconditioned ex vivo. This was performed on the one hand with cytokines (from PBMC culture medium), and on the other hand with general immunostimulating signal(s) [14] and specific tumor-associated antigens (from virus-infected PDAC cell oncolysate vaccine).
PBMC from healthy donors were plated at 1x106 cells/well. Cells were left untreated or were infected with H-1PV or JabCG at MOI10. PDAC cells were plated at 1x106 cells/well in 6-well plates. After 48h PBMCs were washed 3 times with PBS and placed in the transwell system on the top of PDAC cells. The cell survival rates were assessed after 72h using an MTT assay. (A) - PBMCs were left untreated, (B) – PBMCs were treated with IL-2 (100 Units/ml).
Recent work from our group has shown that T3M4 cells are a most appropriate candidate for an allogenic pancreatic cancer vaccine (Angelova et al., manuscript in press), by exhibiting a complete panel of immunogenic cell death determinants upon virus and chemotherapy treatment [15]. Therefore, this cell line was used to induce DC maturation and PDAC antigen presentation after infection with either wt H-1PV or JabCG viruses serving both as adjuvants and oncolytics. Activated dendritic and T-cell populations, cleared from tumor cell debris, were then injected peritumorally in xenograft-bearing mice and neoplastic development was followed for 10 weeks. Three groups of animals were compared, in which PBMCs were primed with mock, H-1PV and JabCG-infected T3M4 cells, respectively. As shown in figure 4, tumor growth was strikingly delayed in both experimental groups, where virus-infected T3M4 cells were used as part of the vaccine preparation protocol. Interestingly, this tumor growth retardation was significantly stronger in the case of JabCG treatment (p<0.05 vs mock and H-1PV). It should be stated that the activating effect of infected T3M4 cells on cocultured PBMCs may be traced back to the fact that parvovirus-induced T3M4 cell death is not only immunogenic, but is also accompanied with some production of progeny virions. Indeed, parvoviruses cause the abortive infection of PBMC subsets, resulting in their activation (see above and [9]). In agreement with this possibility, the expression of proinflammatory cytokines (TNFα, IL-1, IL-6, IL-8) in DCs from pancreatic cancer patients was markedly induced after infection with JabCG and, to a lesser extent, wt H-1PV, as determined by quantitative RT-PCR (Figure 4). A lower but significant induction of IL-12 and type I IFN response factors ISG-15 and OAS, was also achieved by both viruses, whereas IL-10 expression was down-modulated in infected cells.

Figure 4: Comparison of adjuvant effects of H-1PV and JabCG viruses to a PDAC allogenic vaccine.
(A) NOD.SCID mice (n=24) bearing xenografted tumors 5mm3 from two PDAC patients (12 mice per patient) were divided in three groups (n=8) and injected peritumorally with a DC+T-cell mix (5x105+ 3x106) stimulated with T3M4 cells infected respectively as mock, H-1PV or JabCG and after depletion of T3M4 superinfected with the same viruses at MOI5. (B) dendritic cells were obtained from monocytes/PBMCs under GM-CSF (50 ng/ml) and IL-4 (1000U/ml) treatment. Cells were plated at 1x106cells/well in 6-well plates, left untreated or were infected with H-1PV or JabCG at MOI10. 24 hpi cells were collected in RNAeasy buffer and processed for qPCR. The data represent fold induction of gene expression compared to untreated samples. (Number of healthy donors n=12) * - p = 0.05 compared to MOCk.
In a clinical setup, the efficiency of the above PV-assisted therapeutic vaccination protocol may be enhanced by priming the response through a prior intraoperative virus application at tumor site, followed by a course of chemotherapy. This prompted us to use the same animal model to test such a stepwise combinatorial protocol and ensure that chemotherapy (in particular treatment with gemcitabine, the standard drug against PDAC) did not interfere with the protective effect of subsequent reconstitution with ex vivo activated immune cells. As shown in figure 5, the DC/T-cell mixture primed ex vivo with mock-treated T3M4 cells slowed down tumor growth in animals subjected to chemo (gemcitabine)-viro (H-1PV) treatment (CVT) prior to reconstitution, compared to absence of pretreatment (Figure 5). These cells can thus serve as a valuable source of tumor antigens to form the basis for an allogenic PDAC vaccine that is also effective under CVT conditions. Furthermore, exposure of T3M4 cells to CVT strikingly enhanced their capacity for priming the DC/T-cell mixture ex vivo, resulting in the full inhibition of the growth of 2 patient-derived tumorxenografts in reconstituted animals (Figure 5). As such, complete tumor suppression was not achieved when H-1PV alone was used as adjuvant (Figure 4), these data give evidence to suggest that H-1PV and gemcitabine cooperate in enhancing the immunogenicity of PDAC cells. This conclusion was supported by comparing gemcitabine, H-1PV and the combination thereof for their respective abilities to activate immune cells upon treatment of cocultures of T3M4 cells with PDAC patients-derived PBMCs. Stimulation of immune cells was assessed from their cytokine profiles, as determined by qRT-PCR. As shown in Figure 5, transcription of a number of cytokine and type I IFN response genes was enhanced by both viral and drug components of CVT. Interestingly, the virus acted synergistically with gemcitabine to induce IFNγ and, to a lesser extent, IL-2 expression. This synergism was not observed when PBMCs were subjected to CVT in absence of T3M4 cells (data not shown), indicating that the cell vaccine mediated the cooperative effect of both CVT components on at least some facets of immune cell activation.

Figure 5: CVT conditioning of T3M4 cells improves the vaccination effect of T3M4 cells (A) NOD.
SCID mice (n=24) bearing xenografted tumors 5mm3 from two PDAC patients were divided in three groups (n=9) and injected peritumorally with a DC+T-cell mix stimulated with T3M4 cells treated as mock or CVT (H-1PV MOI10 and Gemzar 1.2 ng/ml), (B) PBMCs were obtained from healthy donors (n=10). Cells were plated at 1x106 cells/well in 6-well plates and co-cultured with T3M4 at ratio 10:1. Cells were left untreated, infected with H-1PV at MOI10, treated with Gemzar (1.2 ng/ml) or combined CVT. At 24 hpi cells they were harvested for qPCR. The data represent fold induction of gene expression compared to untreated samples. * - p = 0.05 compared to MOCK (C) Dendritic cells were obtained from monocytes/PBMCs under GM-CSF (50 ng/ml) and IL-4 (1000 U/ml) treatment. Cells were plated at 1x106 cells/well in 6-well plates, left untreated, were infected with H-1PV at MOI10, treated with Gemzar (40 ng/ml) or combined CVT. 24 hpi cells were collected in RNAeasy buffer and processed for qPCR. The data represent fold induction of gene expression compared to untreated samples. (Number of healthy donors n=10).
As CVT can also act in a direct way on immune cells, we also tested the ability of gemcitabine, either alone or in combination with H-1PV, to activate dendritic cells isolated from PDAC patients’ PBMCs. As shown in Figure 5, the gemcitabine/H-1PV combination was effective in inducing transcript accumulation for a number of cytokines (including TNFα and various interleukins) and type I interferon response (OAS) genes.
Arming with CpG motifs enhances the anticancer efficiency of systemically applied PVs. The immunomodulating and therapeutic effects of systemic parvovirus application were evaluated in a previously described orthotopic PDAC model in rats [16]. Animals carrying established pancreatic tumors and hepatic metastatic nodules were treated through a single intravenous application of 1x109 pfu of either wt H-1PV or JabCG. The survival of treated animals was determined, and immunological markers and virus distribution were monitored in rats sacrificed 3 and 10 days after virus administration. As illustrated in Figure 6, the application of both viruses significantly increased the median survival of treated animals (p<0.001). Interestingly, JabCG had a stronger therapeutic potential compared to wt H-1PV (p<0.001). Immune cell infiltration of tumors was evaluated by flow cytometry at both early and late times after virus injection (Figure 6). JabCG induced a more pronounced early influx of CD3, especially activated (CD3+CD25+) and memory T lymphocytes, and NK cells compared to H-1PV. At later stages this effect became more evident for both viruses.

Figure 6: Immunotherapeutic effects of H-1PV and JabCG viruses upon systemic application.
The expression profile of distinct cytokines was determined in the serum of treated animals, revealing that the concentrations of TNFα, IFNγ and IL-2 were enhanced after JabCG injection (Figure 6). Cytokine transcripts were also quantitated by RT-PCR in spleen tissues and in cultures of splenocytes isolated from treated rats. As regards spleen tissues, infection of the animals correlated with both transient detection of viral transcripts, and long-standing induction of TNFα and IFNγ expression (Figure 6). Like sera (Figure 6), spleens from JabCG-infected rats sustained higher levels of cytokine expression than those from wt H-1PV -treated animals. Interestingly, splenocytes isolated from tumor-bearing rats infected with JabCG (vs. wt H-1PV) displayed higher IFNγ production upon in vitro stimulation with direct PV inoculation or with virus-infected tumor cells (Figure 6). Altogether, these results pointed to the enhanced ability of JabCG (in comparison with wt H-1PV) to activate, in tumor-bearing animals, an antitumor cellular immune response.
The sera from control and virus-treated rats were compared for the presence of virus-neutralizing antibodies (α-H-1PV). As expected, at an early time-point (3 days) after infection, none of the sera protected indicator cells from virus infection (data not shown). At a later time (10 days p.i.), neutralizing antibodies were detected in sera from infected but not control animals (Figure 6). Interestingly, the systemic application of JabCG elicited higher α-H-1PV titers compared to the i.v. injection of wt H-1PV.
In summary, JabCG proved to surpass wt H-1PV in its ability to prolong the life of tumor-bearing animals upon single intravenous application. This enhanced anticancer effect of the CpG-armed virus correlated with a greater capacity for inducing (i) the early infiltration of tumors with NKs and activated T-lymphocytes, (ii) the production of cytokines (in particular IFNγ) in spleen, and (iii) elevated titers of circulating α-H-1PV antibodies.
Discussion
This study gives evidence that arming H-1PV with CpG motifs results in superior immunostimulating properties of JabCG not only in rats (the natural host for H-1PV) but also in human in vitro and in vivo reconstituted systems. These adjuvant features correlated with an enhanced capacity of JabCG for rat and human tumor suppression in immunocompetent and reconstituted animal models, respectively. In cultured human PBMCs and DCs, the JabCG virus acted as a general immune stimulator, inducing elevated expression of proinflammatory and Th1 cytokines. Furthermore, co-stimulatory molecules and MHC II were upregulated on JabCG-infected antigen-presenting cells, thereby fulfilling the prerequisites for the development of an antitumor response.
(A) 30 rats bearing 10days old DSL-6A/C1 tumors in the pancreas and a liver nodule were divided in three groups (n=10) and injected i.v. with PBS, H-1PV or JabCG (B) two rats per group were sacrificed and tumor infiltrating lymphocyte subpopupations were evaluated at days 3 and 10 post treatment. Tumors were harvested, treated with collagenase and DNAse and smashed over metal grids. Single cell suspension was stained with corresponding antibodies and signals were measured by FACS. (C) serum level of cytokines measured by ELISA. (D) RT-PCR from tumor tissues at 3 and 10 days after virus i.v. inoculation (E) IFNγ release from splenocytes of mock, H-1PV or JabCG -treated animals in response to infection with H-1PV or JabCG, or co-culture with infected DSL-6A/C1 cells (F) Sera were collected from sacrificed animals and the level of αH-1PV was measured by protection MTT assay. The values represent the percentage of surviving cells compared to mock. * - p = 0.05 compared to mock.
Besides exerting direct effects on immune cells, JabCG was found to act as adjuvant to an allogenictumor cell vaccine against pancreatic cancer. This was revealed through the enhanced capacity of the JabCG-infected vaccine for activating human immune cells ex vivo, in an autologously reconstituted system of patient-derived PDAC xenografts. To serve as an allogenic vaccine, T3M4 cells were selected among a panel of human PDAC cell lines due to the fact that wt H-1PV and the standard anti-PDAC drug gemcitabine both induce their immunogenic cell death (ICD). While their surface is calreticulin-positive, T3M4 cells show a striking increase in extracellular HMGB1 and ATP/IL-1 release upon treatment with H-1PV and the proapoptotic drug gemcitabine (Gemzar®), respectively (Angelova et al., ms in press). The respective induction of these ICD determinants argued for the combinatorial use of H-1PV and Gemzar® as adjuvants. This led us to apply the chemovirotherapy protocol to potentiate the ability of T3M4 cells to serve as allogenic vaccine. This strategy was validated in a preclinical system whereby human immune cells were exposed to the vaccine ex vivo before being used to reconstitute immunodeficient mice bearing autologous PDAC tumors. The prominent IFNγ and IL-2 expression induced by the chemovirotherapy-treated vaccine in PBMC cocultures in vitro was translated into an improved therapeutic outcome in vivo when using this treatment as adjuvant.
In a previous study, the JabCG virus was used as adjuvant to an autologous therapeutic vaccine against lung metastases [4]. In the latter work, the virus was applied ex vivo to the tumor cell vaccine, and its anticancer activity was traced back to the immunological effects of the vaccine, correlating with enhanced IFNγ expression in mediastinaltumor-draining lymph nodes. Similar immunological changes were observed in the present PDAC rat model, where JabCG was administered systemically. Our data argue for the possibility of the i.v. route of parvovirus administration being used to convert an endogenous tumor into an autologous vaccine. This possibility was supported by the comparisons of JabCG and wt H-1PV viruses, which revealed the greater ability of the CpG-armed virus to protect intravenously treated rats against PDAC development. This improved anticancer effect was found to correlate with the higher capacity of JabCG, compared to H-1PV, for inducing hallmarks of immunotherapy responses, i.e. Th1 cytokine profiles in spleen and cultured splenocytes, and early infiltration of tumors with both CD3 and NK cells. Given their cytotoxic properties, the latter cells were likely to participate in the improved therapeutic outcome of JabCG vs. H-1PV intravenous administration. This immune mediation would be in keeping with our previous finding that JabCG cannot be distinguished from H-1PV as far as direct viral oncolysis is concerned [4].
It should also be stated that besides their adjuvant effect on tumor cell vaccines, H-1PV and JabCG viruses exerted general immunostimulating activity as a result of their direct interaction with distinct PBMC subsets, including dendritic cells. We recently reported that while being abortive, PV infection of immune cells can lead to their activation [9]. This was further evidenced in the present study by the induction of various cytokine genes expression in H-1PV and JabCG-infected PBMCs, with a bias towards a Th1 cytokine profile that is indication of cell-mediated immune activation. Cytokines released by parvovirus-infected PBMCs included TNFα and type I interferons which may also contribute to toxicity for target tumor cells [5,9]. In keeping with its arming with immunostimulating motifs, JabCG was superior to its wt H-1PV regarding these direct activating effects on immune cells, which may also reinforce the anticancer potency of the recombinant JabCG virus. In agreement with this possibility, JabCG-infected PBMCs were found to exert a stronger toxic bystander effect on target transformed or tumor cells, as compared with wt H-1PV-infected PBMCs, in a transwellsystem that prevents PBMCs and target cells from direct contact. Since JabCG and H-1PV cannot be distinguished regarding their cytotoxicity and production levels [4], the higher bystander killing effect of JabCG can be assigned to the enhanced ability of that virus to induce production and release of toxic cytokines from PBMCs. A further potential outcome of the direct interaction of parvoviruses with immune cells would be the transport of incoming virions in the blood stream in association with these cells serving as carriers. The concept of using adoptive transfer of normal T-cells loaded with OVs to achieve virus transport and delivery has been recently validated [17]. Our present results showing that infected PBMCs can exert a bystander toxic effect on permissive target cells in a transwell system raises the possibility that besides producing cytokines, infected blood cells may release input virions in the vicinity of tumors, thereby contributing to the induction of viral oncolysis. It should be stated that even under mitogen/IL-2 stimulation conditions, PV infection of PBMCs remains abortive [9], pointing to a possible role of these cells in virus transport instead of de novo production.
The increased titer of virus-neutralizing antibodies upon CpG enrichment of the parvoviral genome represents another facet of the enhanced immunostimulating capacity of JabCG. This feature is especially interesting with respect to antiviral vaccine formulation strategies. Indeed, such a genomic modification could potentially result in an increase in the immunogenicity of any attenuated DNA virus used as live vaccine, while affording the possibility to reduce the amount of toxic adjuvant additives within the formulation. An attractive opportunity constitutes in the use of DNA-based antiviral vaccines.
In conclusion, our data support the use of genomic CpG enrichment of DNA OVs in order to enhance their immunostimulating potential for anticancer therapy. In general, rodent PVs do not trigger overt immune/inflammatory reactions in their natural host, and apparently also in humans, in contrast to some other pathogenic viruses engineered to become oncolytic. This provides a unique opportunity for designing parvovirus-based immunomodulators to fine-tune anticancer immune responses and uncover cryptic tumor antigens. The ongoing clinical study of wt H-1PV in cancer patients [2] should pave the way for putting modified derivatives, including CpG-armed viruses, to the clinical test.
Acknowledgement
This study was supported by a grant from the German Research Council, (DFG - GI 802/1-1, RA 1891/2-1). The authors would like to thank Prof. P. Beckhove, Mariana Bucur (Translational Immunology, DKFZ-Heidelberg) for collaboration on DC+T-cell system, Sven Rüffer, BrunhildeBentzinger and Monika Meinhardt (UniKlinik Heidelberg) for excellent assistance.
References
- Rommelaere J, Geletneky K, Angelova AL, Daeffler L, Dinsart C, et al. (2010) Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev 21: 185-195.
- Geletneky K, Huesing J, Rommelaere J, Schlehofer JR, Leuchs B, et al. (2012) Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastomamultiforme: ParvOryx01 protocol. BMC Cancer 12: 99.
- Grekova SP, Aprahamian M, Daeffler L, Leuchs B, Angelova A, et al. (2011) Interferon gamma improves the vaccination potential of oncolytic parvovirus H-1PV for the treatment of peritoneal carcinomatosis in pancreatic cancer. Cancer Biol The 12: 888-895.
- Raykov Z, Grekova S, Leuchs B, Aprahamian M, Rommelaere J (2008)Arming parvoviruses with CpG motifs to improve their oncosuppressive capacity. Int J Cancer 122: 2880-2884.
- Raykov Z, Grekova SP, Hörlein R, Leuchs B, Giese T, et al. (2013) TLR-9 contributes to the antiviral innate immune sensing of rodent parvoviruses MVMp and H-1PV by normal human immune cells. PLoS One 8: e55086.
- Morales O, Richard A, Martin N, Mrizak D, Sénéchal M, et al. (2012) Activation of a helper and not regulatory human CD4+ T cell response by oncolytic H-1 parvovirus. PLoS One 7: e32197.
- Moehler MH, Zeidler M, Wilsberg V, Cornelis JJ, Woelfel T, et al. (2005) Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum gene ther 16: 996-1005.
- Bhat R, Dempe S, Dinsart C, Rommelaere J (2011) Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int J Cancer 128: 908-919.
- Grekova S, Aprahamian M, Giese N, Schmitt S, Giese T, et al. (2010) Immune cells participate in the oncosuppressive activity of parvovirus H-1PV and are activated as a result of their abortive infection with this agent. Cancer BiolTher10: 1280-1289.
- Raykov Z, Grekova S, Galabov AS, Balboni G, Koch U, et al. (2007) Combined oncolytic and vaccination activities of parvovirus H-1 in a metastatic tumor model. Oncol Rep 17: 1493-1499.
- Raykov Z, Savelyeva L, Balboni G, Giese T, Rommelaere J, et al. (2005) lymphocytes and myeloid dendritic cells in lymphoid organs are preferential extratumoral sites of parvovirus minute virus of mice prototype strain expression. J Virol 79: 3517-3524.
- Adair RA, Roulstone V, Scott KJ, Morgan R, Nuovo GJ, et al. (2012) Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Science translational medicine 4: 138ra177.
- Grekova SP, Raykov Z, Zawatzky R, Rommelaere J, Koch U (2012) Activation of a glioma-specific immune response by oncolytic parvovirus Minute Virus of Mice infection. Cancer Gene Ther 19: 468-475.
- Schmitz-Winnenthal FH, Escobedo LV, Beckhove P, Schirrmacher V, Bucur M, et al. (2006) Specific immune recognition of pancreatic carcinoma by patient-derived CD4 and CD8 T cells and its improvement by interferon-gamma. International journal of oncology 28: 1419-1428.
- Kroemer G1, Galluzzi L, Kepp O, Zitvogel L (2013) Immunogenic cell death in cancer therapy. Annu Rev Immunol 31: 51-72.
- Angelova AL, Aprahamian M, Grekova SP, Hajri A, Leuchs B, et al. (2009) Improvement of gemcitabine-based therapy of pancreatic carcinoma by means of oncolytic parvovirus H-1PV. Clin Cancer Res 15: 511-519.
- Qiao J, Kottke T, Willmon C, Galivo F, Wongthida P, et al. (2008) Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolyticvirotherapy and immunotherapy. Nature medicine 14: 37-44.
- Raykov Z, Balboni G, Aprahamian M, Rommelaere J (2004) Carrier cell-mediated delivery of oncolytic parvoviruses for targeting metastases. Int J Cancer 109: 742-749.
- Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, et al. (2006) An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res 12: 4652-4661.
- Li J, Bonifati S, Hristov G, Marttila T, Valmary-Degano S, et al. (2013) Synergistic combination of valproic acid and oncolytic parvovirus H-1PV as a potential therapy against cervical and pancreatic carcinomas. EMBO molecular medicine 5: 1537-1555.
Copyright: © 2014 RaykovZ. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.