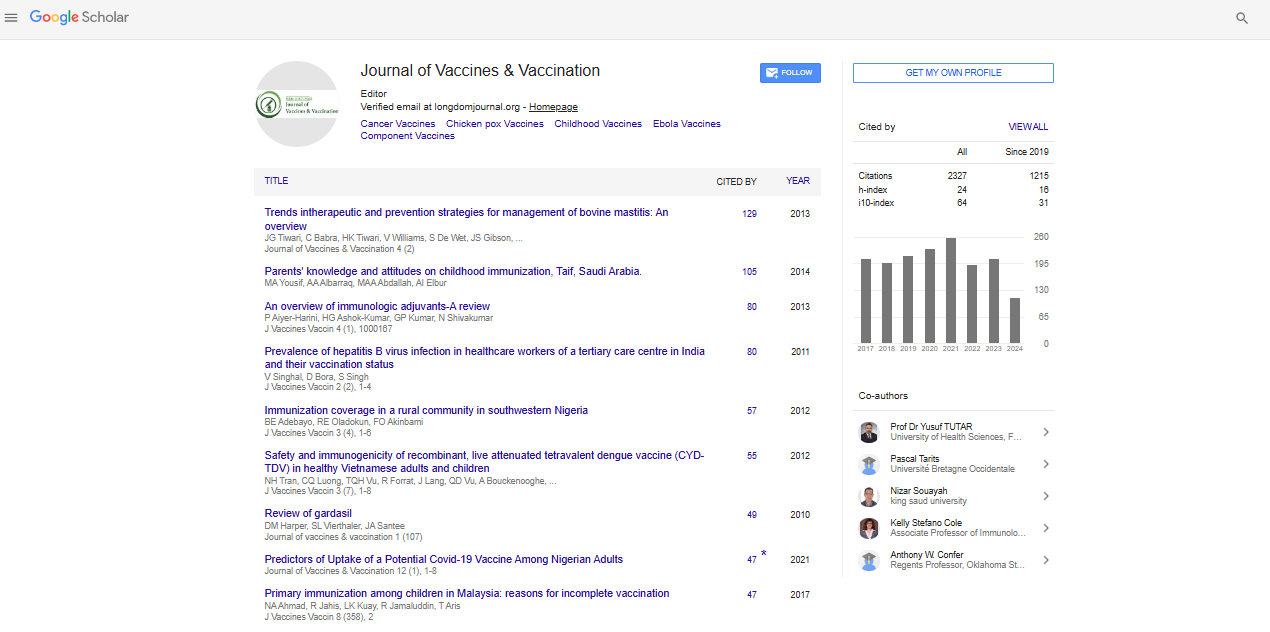

Indexed In
- Academic Journals Database
- Open J Gate
- Genamics JournalSeek
- JournalTOCs
- China National Knowledge Infrastructure (CNKI)
- Scimago
- Ulrich's Periodicals Directory
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- MIAR
- University Grants Commission
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2022) Volume 13, Issue 1
Genetic Analysis of SARS-CoV-2 and the Common Golden Nucleotides to Human Gene
Hamed Babaee*Received: 03-Jan-2022, Manuscript No. JVV-22-14933; Editor assigned: 05-Jan-2022, Pre QC No. JVV-22-14933 (PQ); Reviewed: 21-Jan-2022, QC No. JVV-22-14933; Revised: 26-Jan-2022, Manuscript No. JVV-22-14933 (R); Published: 02-Feb-2022, DOI: 10.35248/2155-9627.22.13.471
Abstract
Background: The COVID-19 pandemic broke out in Wuhan, China in 2019. It still persists in 2021, for different strains of the novel coronavirus have appeared; therefore, new solutions and treatments are necessary.
Methods and results: This study aimed to analyze the COVID-19 from a different angle by conducting a genetic analysis on data of 24 SARS-CoV-2 samples from different countries in alignment with each other as well as the Wuhan reference virus and the human genome. The analysis helped identify genetic differences in viruses and find a unique 17-nucleotide sequence between human genes, viruses, and enzymes. The results can be employed to determine the onset and progression of the disease.
Conclusion: This sequence can be involved in DNA replication and production of new proteins. Its alignment with the EPPK1 gene can cause various symptoms.
Keywords
COVID-19; SARS-CoV-2; RNA sequencing
Abbrevations
SARS-CoV-2: Severe Acute Respiratory Syndrome Coronavirus-2; ACE2: Angiotensin-Converting Enzyme 2; ORFs: Open Reading Frames.
Introduction
In 2019, the world was struck by a novel viral contagious disease called the COVID-19 or the coronavirus disease 2019. Apparently, this disease originated from a seafood wholesale market in Wuhan, China with an unknown zoonotic origin. At first, China had the highest case report and mortality rates, which were then mitigated by severe quarantine measures. Nearly every country has experienced an epidemic of this virus ever since. By April 25, 2021, nearly 147 million cases were reported worldwide. The highest case reports were recorded in the USA, India, Brazil, France, Russia, Turkey, England, Italy, Spain, and China. In fact, China is known as the possible origin of the COVID-19 ranked 95th at https://worldometers.info without any mortality. Urban and national population densities, safety, epidemic management, viral strains, and population genetics affect the variations of epidemic statistics and mortality rates. Moreover, the novel coronavirus has undergone separate mutations in different countries and has probably adapted to different population genetics of different areas based on its nature; thus, it has found new ways of replicating and initiating new conditions in different countries.
Many detection and treatment methods have been analyzed and evaluated. Most of them are based on clinical imaging. Several companies have produced globally available vaccines tested in clinical trials. Some of these vaccines only have positive results in preliminary trials, whereas some others have major and minor side effects on different people. Research on vaccines and drugs is still the work in progress.
SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2) is an RNA positive single-strand virus. Generally, it enters human cells by connecting to the Angiotensin-Converting Enzyme 2 (ACE2) [1,2]. In addition to the connection and entry of the virus into human cells, its replication and pathogenesis are still comprehended partially. Two possible routes might be taken by the virus after entering the body. It might directly engage in the production of pathogenic proteins in specific organs or indirectly connect its viral sequence segments to human sequences and genes. The virus uses and transcribes the human genes to change cellular functions or produce new proteins in order to invade cells and tissues. In fact, the virus depends completely on a host like humans; thus, it is difficult to develop a virus in laboratory conditions, unlike rhinoviruses. Since the coronavirus genome acts directly as an mRNA, it needs neither splicing nor transcription for translation. Therefore, it can be translated without an intermediate.
When the RNA of the virus is released into the cell, it moves directly to ribosomes on the endoplasmic reticulum without needing to enter the nucleus to be transcribed. Therefore, it directly produces its antigens [3]. This virus might also begin its invasive activities after it is fixed into a secure position and is protected within the organism.
The SARS-CoV-2 genome has 10 Open Reading Frames (ORFs). Its structural proteins E, N, M, and S along with other accessory proteins are coded into the ORFs 2-10. Proteins E, M, and S shape the virus coating, whereas the N protein helps pack its RNA genome. The NSP proteins are a group of functional proteins involved in host infection, virus replication, RNA transcription, splicing and modification, protein translation, and synthesis. Proteins like PLpro 3, CLpro, RdRp, and Nsp13-helicase have biologic functions and vital enzyme active sites, which make them important targets. NSP1, NSP3C, and ORF7a of the novel coronavirus disrupts a host’s innate immune system and helps with the evasion of the virus immune system [4]. These proteins accelerate the viral infiltration by disrupting a host system’s defense system. According to different research findings, several viral proteins attack the 1-β hemoglobin chain and remove the iron from the porphyrin, a process which reduces the oxygen transfer capabilities of the hemoglobin [5]. Additionally, some studies indicated the hormonal status had a significant role in higher mortality rates, something which explains higher mortality rates and disease intensity in men than in women [6].
Materials and Methods
In this study, 24 FASTA sequences of the SARS-CoV-2 were analyzed and evaluated from 21 countries. The USA, Iran, and China, Each had two sequence samples. All data was downloaded through specific accession numbers within the NCBI Nucleotide database with deposit dates from April 2020 up to March 2021.
CLC Genomics Workbench was employed in this experiment to align the sample sequences and the human genome for analysis and assessment of differences and commonalities in order to finally draw the phylogenetic tree. The lengths of sequences ranged between 29003 bp and 29903 bp. The alignment and evaluation of sequences were based on the Wuhan Reference Virus Genome (NC_045512.2), which was the longest viral genome. Therefore, to facilitate the identification of nucleotide mutation locations, alignment position order counting was done for the sake of uniformity. Moreover, a single identification method was adopted, although the identification region of each virus mutation can easily be observed from its sequence. Usually, differences in sequences lengths are nearly 70 bp in the 5´ and 3´ ends, whereas the ORFs 1-10 are perfectly aligned, except for rare mutations.
Results
The research samples included different strains ranging from two (France and USA1 variants) up to 853 (China 2 variant) mutations. All samples, except Wuhan and the USA1 variants, had unique and specific mutations. Mutations were given different names such as “T 241 C/T”. The first letter T or D stands for the mutation type, i.e., translocation or deletion. The following digits indicate the base number of the mutation within the alignment position order, whereas C/T denotes the substitution of cytosine for thymine.
The South African and Brazilian (i.e., MT324062.1 and MT350282.1) samples resemble the Wuhan variant in length (29903 bp); however, the South African variant had three translocation mutations separating it from the Wuhan variant. The Brazilian variant had ten translocations, four of which resembled the Wuhan variant. Three C/T mutations were at positions 241, 3037, and 14408 of the alignment positioning (T 241 C/T, T 3037 C/T, and T 14408 C/T) and a single A/G substitution (T 23403 A/G). Out of all these variants, only the samples from France, South Africa, and Turkey had unique mutations.
Four translocation mutations (T 241 C/T, T 3037 C/T, T 23403 A/G, and T 14408 C/T) of the Wuhan virus were observed in ten countries: Iran, Colombia, Italy, Nepal, Vietnam, India, England, South Korea, and Brazil. This probably indicates that these viruses originated from Wuhan, whereas the others are missing these mutations.
The sequences of variants from England, Belarus, the Philippines, the USA 2, and China 2 had deletion mutations in addition to translocation mutations. Interestingly, the viruses containing deletions have been detected in 2021.
The English variant had a 24-nucleotide deletion (D 23598-23621) in gene S. The Belarus virus had two mutations including a 9-nucleotide deletion (D 686-694) in ORF1ab and a 15-nucleotide deletion (D 27764-27778) in ORF7b. The Philippines virus had three mutations including a 9-nucleotide deletion (D 11288-11296) in ORF1ab, a 6-nucleotide deletion in (D 21766-21771) in ORF7b, and a 3-nucleotide deletion (D 21994-21996) in gene S. The USA variant 2 had a 10-nucleotide deletion (D 80-89) in the 5’, whereas the China variant 2 had five long deletions: a 26-nucleotide deletion (D 27375-27400) in ORF6, a 130-nucleotide deletion (D 27416-27545) in ORF7a, a 332-nucleotide deletion (D 27555- 27886) in ORF7a and ORF7b, a 104-nucleotide deletion (D 27908- 28011) in ORF8, and a 233-nucleotide deletion (D 28023-28255) in ORF8. An interesting feature of the China variant 2 is the high density of deletions that removed ORF7b, ORF7a, and ORF8 nearly completely. No samples had a deletion in ORF10, gene N, gene M, gene E, or ORF3a. More than 50% of mutations in 2020 were in ORF1ab, whereas the 2021 samples had less than 50% of the mutations in ORF1ab. Furthermore, the number of mutations in 2021 has been 2 to 3 times further than the average number in 2020. Despite all these mutations, the virus continues to function and has not lost its aggressiveness.
The phylogenetic tree (Figure 1) demonstrates differences between the China virus 2 and the Wuhan virus at the beginning and end of the graph within the 2019-2021 periods. Accordingly, the Wuhan variant is the origin and is shown as a subordinate of several viruses. Additionally, the China virus 2 is a completely new variant compared to all other viruses in other branches of the graph with the Philippines virus on a corresponding branch having different mutations. According to the recent mortality rates reported in China and the high mutation rates observed in China variant 2 with the deletions of ORF7b, ORF7a, and ORF8, these genes can be considered very important.

Figure 1: Design of cropland sites along with preciptation gradiens from east to west in Jilin.
Mutations in the functional protein PLpro also exist within the South African variant alignment position sequence (T 5572 T/G), the Columbia variant (T 5298 N/C), the Philippines variant (T 4964 G/A and T 5388 A/C), and the China 2 variant (T 5653 C/T).
The RdRp protein is mutated in the Indian variant (T 14408 C/T and T 16176 C/T) Vietnam, Nepal, Italy, Columbia, Iran 1, Brazil, Iran 2, Wuhan, England, and South Korea variants (T 14805 T/C) as well as the Belarus variant (T 15372 T/G), the Philippines variant (T 14676 T/C, T 15279 T/C), and the China 2 variant (T 15165 G/A).
The Nsp13-helicase functional protein is mutated in the Columbia variant (T 17470 T/C), the Brazil variant (T 17247 C/T), the Philippines variant (T 17615 G/A), and the USA 2 variant (T 17014 T/G).
The Nsp1 is also mutated in the Spain variant (T 313 T/C) and the France variant (T 618 G/A). ORF7a (27759-27394) is also mutated in the Belarus variant (T 27670 T/G), the China 2 variant (T 27412, T 27410 C/T, T 27407 C/T, T 27405 A/T, D 2755-27759, T 27553- 27554 C/T, D 27416-27545, and T 27413 A/T).
Considering the increase in mutation and contagion, the researchers sought further effective interacting elements because the virus genome must interact with other parts of the human genome for replication and pathogenesis and use the human genome for invasive purposes.
In the next part of the analysis, RNA sequencing was employed to align 24 virus sequences with the HG38 human genome. According to the results, a small sequence was similar to the human genome in the genome sequence of all viruses. It aligned to the EPPK1 gene in chromosome 8. This 17-nucleotide sequence (TCCTGCTGCAGATTTGG) containing the PstI restriction site is the unique sequence between all SARS-CoV-2 sample genomes and the hg38 human genome. The sequence in all 24 sample genomes from different countries in gene N of the SARS-CoV-2 virus was nearly in 28500-29500 positions and 29474-29458 positions of the virus sequence alignment position. This sequence is also located in the EPPK1 gene at positions 3961-3977 (Figure 2).

Figure 2: The unique sequence of 17 nucleotides (TCCTGCTGCAGATTTGG) in the 24 SARS-Cov-2 genes collected from different countries and the EPPK1 gene.
Discussion
PstI is a type-II endonuclease restriction enzyme. Its structure consists of two components, one of which is a restriction enzyme that cuts external DNA, whereas the other one is a methyltransferase that protects endogenous DNA with the histone methylation to combine both defense mechanisms against the invading virus [7]. Hypothetically, this virus has reverse-engineered these endonuclease abilities to protect itself. PstI is useful for replicating DNA because it creates a selective system for producing combinatorial DNA molecules [8]. Probably, the coronavirus functions cooperatively with the EPPK1 and PstI genes to produce different novel proteins causing some people to have no or slight symptoms. However, some patients have popular COVID-19 symptoms such as a fever, dry coughs, fatigue, different pains, diarrhea, conjunctivitis, loss of smell and taste, skin rashes, acute respiratory symptoms, and loss of speech and movement.
Figure 3 depicts that a significant number of coronavirus symptoms were observed in protein expressions and the EPPK1 gene RNA. Accordingly, the expressions of protein and RNA are high in the digestive tract, muscle tissues, lungs, kidneys, bladder, endocrine system, skin, bone marrow, and lymph tissues. This indicates a significant relationship between the COVID-19 and the expression of the EPPK1 gene in the human body. Additionally, the EPPK1 expression is higher in men’s tissues than in women’s, something which follows the higher mortality trend in men.

Figure 3: Protein and RNA expression of EPPK1 gene.
EPPK1 is a known human epidermis auto antigen that cooperates with Plakin genes in the attachment and anchoring of skeletal fibers with the plasma membrane. The Plakin family is a multipurpose organizer of cytoskeleton architecture. It has also been found that they play a role in the uniformity of muscle cells. It was recently discovered that they also exist in the nervous system.
The alignment of the PstI restriction enzyme and the 17-nucleotide region of EPPK1 with the viral sequence might represent a tool or a dangerous weapon based on DNA cloning to use the virus for the disruption of the cellular skeletal system, the disintegration of muscle cells, and the manipulation of the nervous system leading to the COVID-19 symptoms.
Conclusion
Many treatment methods have been used for to cure patients with the COVID-19. Some of these methods are centered around viral functions and proteins, whereas others are based on host receptors and proteins to prevent virus attachment, replication, and pathogenicity. All these methods have certain side effects or fail to overcome the novel variants. Apparently, the human immune system has engineered some methods for fighting the epidemics caused by the new strains. This study addressed the similarities between the human genome and the virus to find possible explanations for the causes of pathogenesis. Undoubtedly, the expressions of any phenotypes whether in viruses or humans as the superior living creatures stem from their genetic sequences and the interaction of their genetic products with other organisms. Every problem must first be considered from a genetic point of view, for the design and engineering of life are summed up in the genetic materials, on which the minds, characters, and behaviors of beings and their responses to different stimuli are based on.
Acknowledgement
Thanks to the Creator of the universe.
Thanks to the NCBI Database for the free sharing of coronavirus data used in this study.
Thanks to Sara Tavakoli Dinani
Data availability
Data analyzed in this study are available in the NCBI’s Nucleotide database.
Data accession code in the GeneBank ( https://www.ncbi.nlm.nih. gov/nuccore/):
MT263074.1 MT050493.1 MT192772.1 MT072688.1 MT324062.1 MT066156.1
MT256924.2 MT320891.2 MT350282.1 MT359866.1 MT320538.2 MT447177.1
NC_045512.2 MT344948.1 MT304475.1 MW059036.1 MW306668.1 MW321432.1
MW633517.1 MW674675.1 MW735441.1 MW796991.1 MW691153.1 MW822593.1
Code availability
(Software application) The CLC Genomics Workbench software (Evaluation version).
REFERENCES
- Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5(4):562-569.
[Crossref], [Google Scholar], [PubMed]
- Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270-273.
[Crossref], [Google Scholar], [PubMed]
- Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10(5):766-788.
[Crossref], [Google Scholar], [PubMed]
- Shirani K, Sheikhbahaei E, Torkpour Z, Nejad MG, Moghadas BK, Ghasemi M, et al. A narrative review of COVID-19: the new pandemic disease. Iran J Med Sci. 2020;45(4):233.
[Google Scholar], [Crossref], [PubMed]
- Zhang Y. Encyclopedia of global health. Sage; 2008.
[Crossref]
- Cascella M, Rajnik M, Aleem A, Dulebohn S, Di Napoli R. Features, evaluation, and treatment of coronavirus (COVID-19). StatPearls. 2021.
[Crossref], [Google Scholar]
- Walder RY, Hartley JL, Donelson JE, Walder JA. Cloning and expression of the Pst I restriction-modification system in Escherichia coli. Proc Natl Acad Sci. 1981;78(3):1503-1507.
[Crossref], [Google Scholar], [PubMed]
- Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW, et al. Construction and characterization of new cloning vehicle. II. A multipurpose cloning system. Gene. 1977;2(2):95-113.
[Crossref], [Google Scholar], [Pubmed]
Citation: Babaee H (2021) Genetic Analysis of SARS-CoV-2 and the Common Golden Nucleotides to Human Gene. J Vaccines Vaccin. 13:471
Copyright: © 2021 Babaee H. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.