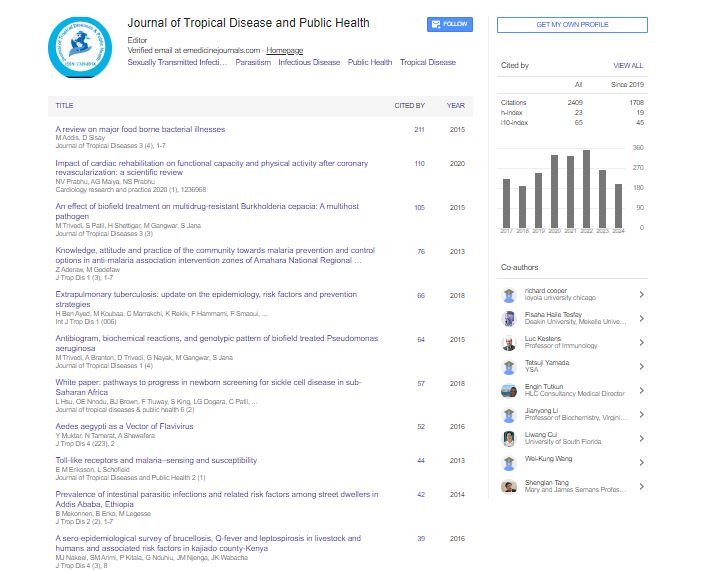

Indexed In
- Open J Gate
- Academic Keys
- ResearchBible
- China National Knowledge Infrastructure (CNKI)
- Centre for Agriculture and Biosciences International (CABI)
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- CABI full text
- Publons
- Geneva Foundation for Medical Education and Research
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Opinion Article - (2022) Volume 10, Issue 6
Evolution of Tuberculosis and its Recombinant Variants
Donald Xiaoge*Received: 24-May-2022, Manuscript No. JTD-22-17345; Editor assigned: 27-May-2022, Pre QC No. JTD-22-17345 (PQ); Reviewed: 10-Jun-2022, QC No. JTD-22-17345; Revised: 17-Jun-2022, Manuscript No. JTD-22-17345 (R); Published: 27-Jun-2022, DOI: 10.35241/2329-891X.22.10.334.
Description
Tuberculosis was responsible for the death of one out of five adults in Europe and North America during seventeenth and nineteenth centuries and even today it remains a leading cause of inflated morbidity and mortality in the developing world.
Infectious diseases affecting humans can be bifurcated into two categories:
•Crowd disease: Crowd diseases are highly virulent and require increased host population densities so that pathogen transmission can be maximized while minimizing the risk of extinction. Emergence of Crowd diseases is often correlated with the enhanced animal domestication practices that helped in the zoonotic transfer of the new pathogens as well as it is also attributed to innovative agriculture practices that led to the increased population densities during Neolithic Demographic Transition Phase which started 10,000 years ago.
•Primitive human disease: Primitive human infections were associated with slow progression to disease and in some cases reactivation after a long period of asymptomatic or latent infection, which are hypothesized as the adaptation of the causal organism to the low population densities so as to replenish the susceptible organism’s reservoir.
Tuberculosis shows the characteristics of both i.e. it can kill 50% of infected population if left untreated and aerosol mode of transmission to infect high population densities. However, tuberculosis also has latency and reactivation propensities which are characteristics of pre-NDT diseases. Before the advent of sequencing techniques, it was believed that Human TB originated from animals but decoding of genomes and phylogenetic analysis have revealed that it is exactly the vice versa of what was hypothesized earlier. Strains that cause tuberculosis in animals diverged from human strains even before Non Destructive Testing (NDT) phase. It has also been found that Mycobacterium Tuberculosis Complex (MTBC) associated with humans has no animal reservoir, further strengthening the humans to animal transmission theory. The MTBC is a group of Mycobacterium species that are genetically similar and comprises of Mycobacterium tuberculosis, Mycobacterium africanum, Mycobacterium canettii, Mycobacterium caprae, Mycobacterium pinnipedii, Mycobacterium microti and Mycobacterium bovis. M. tuberculosis is the most infamous member of the MTBC group registering its presence in one third of the total human population. Mycobacterium canettii and Mycobacterium africanum can also cause tuberculosis in humans and generally infects African patients or African ancestry. Mycobacterium bovis is the broad spectrum pathogen affecting humans, goats as well as bovines. A laboratory mutant strain of Mycobacterium bovis which was isolated by Albert Calmette and Camelle Guerin and known by the name of M. bovis var BCG is the only approved vaccine for TB prevention in children. Mycobacterium caprae infects goats only. Mycobacterium microti infects rodents and also causes disease in immune-compromised patients and Mycobacterium pinnipedii infects seals. It is suggested that MTBC members shared a common ancestor and successive DNA insertions/ deletions resulted in the present speciation among MTBC and also the differences in the pathogenicity was the outcome of the same. Genomic analysis identified about 14 regions called as Region of differences (RD 1-14) which are present in the laboratory strain and are absent in the vaccine strain Mycobacterium bovis var BCG. These deletions helped to understand the pathogenicity related genes.
Conclusion
On the contrary, there are six regions that were found to be deleted from Mycobacterium tuberculosis H37Rv genome as compared to other members of MTBC. They are known by the name “H37Rv deletion 1 to 5 (RvD 1-5)” and “Mycobacterium tuberculosis specific deletion 1 (TbD1)”. Mycobacterium canettii contains all the regions that are found to be deleted in other strains i.e. TbD1, RvD and RD, and is considered to be phylogenetically more similar to the ancestor bacilli. Mycobacterium africanum strains from West Africa do not have RD9 region and strains from East Africa lack RD3 region. Mycobacterium microti do not have regions RD7-10. The commonly found Mycobacterium bovis or what we may call “The classical M. bovis”, which is found in Netherland, Argentina, Great Britain and Spain, possesses highest number of deletions. RD4-10, RD12 and RD13 are found to be deleted. M. bovis var BCG apart from the regions deleted in M. bovis also lacks the regions RD1-RD2 and RD14. These deletions would have occurred after and during attenuation.
Citation: Xiaoge D (2022) Evolution of Tuberculosis and its Recombinant Variants. J Trop Dis. 10:334.
Copyright: © 2022 Xiaoge D .This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.