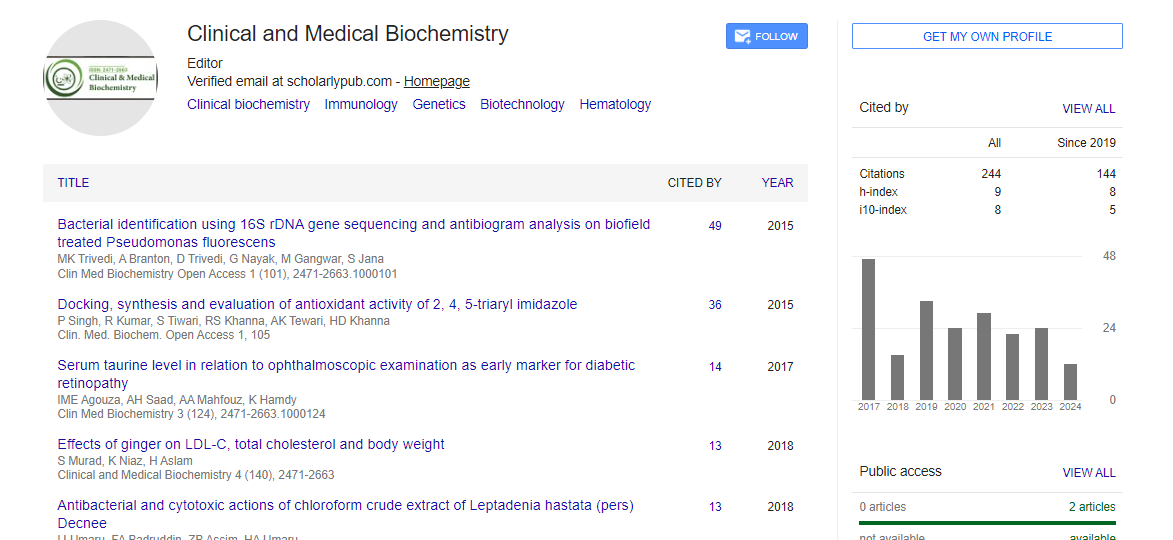

Indexed In
- RefSeek
- Directory of Research Journal Indexing (DRJI)
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Scholarsteer
- Publons
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Perspective - (2023) Volume 9, Issue 6
Enzyme Deficiencies and Dietary Dilemmas: Living with Inborn Errors of Metabolism
Isla Walker*Received: 23-Oct-2023, Manuscript No. CMBO-23-24113; Editor assigned: 26-Oct-2023, Pre QC No. CMBO-23-24113 (PQ); Reviewed: 14-Nov-2023, QC No. CMBO-23-24113; Revised: 21-Nov-2023, Manuscript No. CMBO-23-24113 (R); Published: 28-Nov-2023, DOI: 10.35841/2471-2663.23.9.197
Description
Inborn Errors of Metabolism (IEM) represent a group of rare genetic disorders that disrupt the body's normal metabolic processes. These disorders, often present from birth, result from defects in specific genes encoding enzymes responsible for various metabolic pathways. Considerate the intricacies of inborn errors of metabolism are important for early diagnosis, management, and on-going research aimed at developing effective therapeutic interventions.
Metabolism and its complexity
Metabolism is the intricate set of biochemical processes that occur within cells to maintain life. These processes involve the conversion of nutrients from food into energy, the synthesis of essential molecules, and the elimination of waste products. Enzymes, which act as biological catalysts, play a central role in facilitating these metabolic reactions. In individuals with inborn errors of metabolism, a genetic mutation leads to the deficiency or malfunction of a specific enzyme. This disruption impairs the normal course of metabolic reactions, leading to the accumulation of toxic substances or deficiencies in essential molecules. The consequences of these disruptions can manifest in a wide range of symptoms affecting various organs and systems.
Types of inborn errors of metabolism
Phenylketonuria (PKU): PKU results from a deficiency of the enzyme phenylalanine hydroxylase, which is responsible for converting the amino acid phenylalanine into tyrosine. Without this enzyme, phenylalanine accumulates to toxic levels, leading to intellectual disabilities, developmental delays, and other neurological symptoms.
Maple Syrup Urine Disease (MSUD): MSUD involves a deficiency in the enzymes responsible for breaking down the amino acids leucine, isoleucine, and valine. The accumulation of these amino acids can lead to a distinctive odour resembling maple syrup in bodily fluids, along with neurological symptoms.
Gaucher disease: Gaucher disease results from a deficiency of the enzyme glucocerebrosidase, leading to the accumulation of a fatty substance called glucocerebroside. This build-up primarily affects the spleen, liver, and bone marrow, causing symptoms such as an enlarged spleen and liver, anaemia, and bone abnormalities.
Medium-Chain Acyl-coA Dehydrogenase (MCAD) deficiency: MCAD deficiency hinders the breakdown of medium-chain fatty acids for energy. During periods of fasting or illness, individuals with MCAD deficiency may experience severe metabolic crises, leading to symptoms like vomiting, lethargy, and seizures.
Diagnosis and newborn screening
The diagnosis of inborn errors of metabolism has significantly benefited from advancements in genetic testing and newborn screening programs. Newborn screening, a routine procedure performed shortly after birth, involves the analysis of blood samples to detect metabolic disorders early, even before symptoms appear. Early diagnosis allows for timely intervention and management strategies to prevent or mitigate the adverse effects of these disorders.
Management and treatment
Management strategies for inborn errors of metabolism often focus on dietary modifications, enzyme replacement therapies, and supportive care. For example: In conditions like PKU, strict adherence to a low-phenylalanine diet is important. Specialized formulas and dietary restrictions help control the accumulation of toxic substances, allowing affected individuals to lead relatively normal lives. Some inborn errors of metabolism can be treated with enzyme replacement therapy. This involves administering the deficient enzyme directly to compensate for the genetic deficiency. Enzyme replacement therapies have shown success in certain lysosomal storage disorders. On-going research is exploring the potential of gene therapy to address the root cause of inborn errors of metabolism. By introducing functional copies of the defective genes, gene therapy aims to restore normal enzyme function and metabolic processes.
Challenges and future directions
Despite progress in diagnosis and management, inborn errors of metabolism pose significant challenges. Many of these disorders lack curative treatments, and management often requires a lifelong commitment to dietary restrictions or enzyme replacement therapies. The rarity and heterogeneity of these disorders also present challenges for researchers and healthcare providers. Collaborative efforts between clinicians, researchers, and patient advocacy groups are essential to advance our kind of these disorders, identify new therapeutic targets, and improve outcomes for affected individuals.
Conclusion
Inborn errors of metabolism exemplify the intricate relationship between genetics and biochemical processes essential for life. As our consideration of these disorders deepens, so does the potential for innovative therapeutic interventions. Early diagnosis through newborn screening and ongoing research into gene therapies show belief for improved outcomes and quality of life for individuals affected by these rare genetic disorders. While challenges persist, the progress made in the field of inborn errors of metabolism underscores the importance of continued research and collaboration to unlock new possibilities for diagnosis, treatment, and ultimately, a brighter future for those impacted by these complex genetic conditions.
Citation: Walker I (2023) Enzyme Deficiencies and Dietary Dilemmas: Living with Inborn Errors of Metabolism. Clin Med Bio Chem. 9:197.
Copyright: © 2023 Walker I. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.