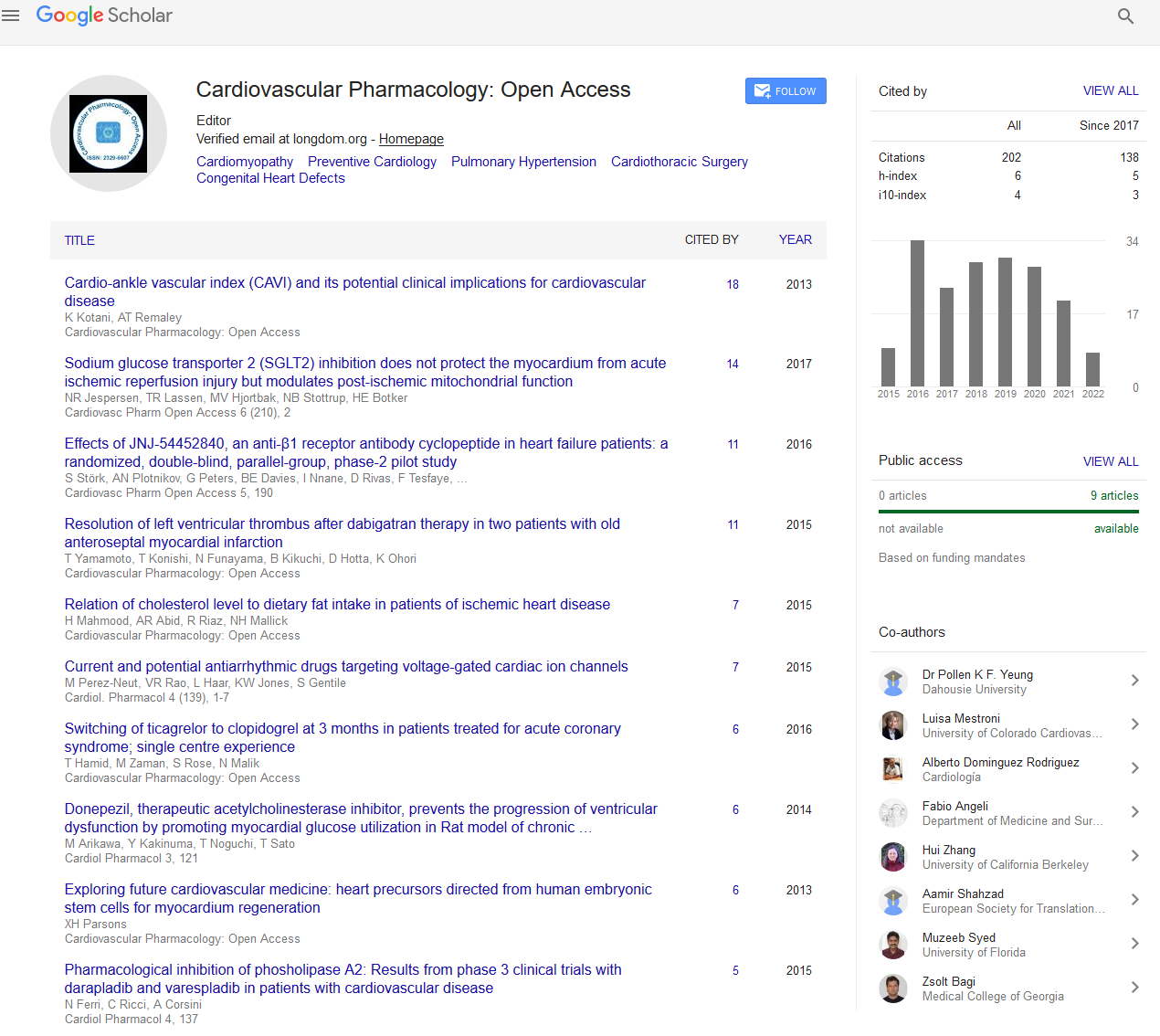

Indexed In
- Open J Gate
- Cosmos IF
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
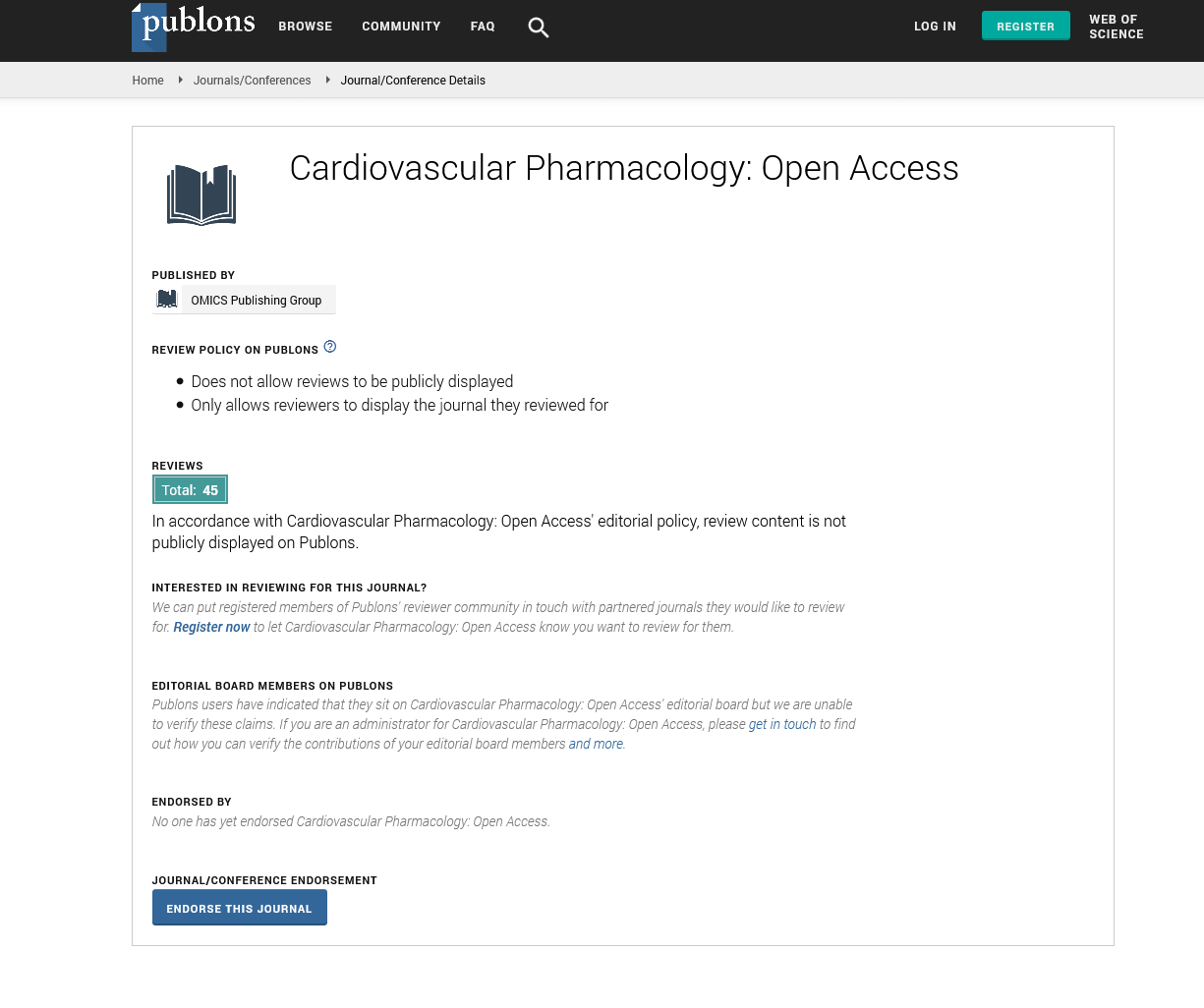
- Publons
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Commentary - (2024) Volume 13, Issue 3
Cardiac Pharmacological Aspects Involved in Fabry Disease
James Carter*Received: 30-Aug-2024, Manuscript No. CPO-24-27832; Editor assigned: 02-Sep-2024, Pre QC No. CPO-24-27832 (PQ); Reviewed: 16-Sep-2024, QC No. CPO-24-27832; Revised: 23-Sep-2024, Manuscript No. CPO-24-27832 (R); Published: 30-Sep-2024, DOI: 10.35248/2329-6607.24.13.401
Description
Fabry disease is a rare X-linked genetic disorder caused by deficiency of the enzyme α-galactosidase A, leading to accumulation of Globotriaosylceramide (Gb3) in various tissues, particularly affecting vascular endothelial cells, renal tubules and cardiomyocytes. The complex pathophysiology of Fabry disease has important implications for cardiovascular health and understanding the available pharmacological interventions is an important area of research and clinical application. This article explains the pharmacological aspects of the cardiac manifestations of Fabry disease, exploring its historical background, notable contributors and therapeutic and adverse effects.
Historically, Fabry disease was first described in 1898 by the Austrian physician Johannes Fabry, who noted the distinctive skin lesions associated with the disease. Over the following decades, it became clear that Fabry disease had systemic manifestations, particularly affecting the heart. The first significant advances in understanding cardiac damage occurred in the late 20th century, when echocardiographic studies began to reveal the extent of Left Ventricular Hypertrophy (LVH) and other structural changes in the heart in affected individuals. These results were essential in establishing the link between enzyme deficiency and cardiovascular complications.
From a pharmacological perspective, Enzyme Replacement Therapy (ERT) has been the mainstay of treatment since its introduction in the late 2000s. Agalsidase β and agalsidase α are two recombinant human forms of FFA that have been shown to be effective in reducing Gb3 deposition and alleviating symptoms associated with Fabry disease. Clinical studies have shown that TES can lead to LVH regression and improvement in left ventricular function, which is essential for improving quality of life and reducing morbidity in affected individuals. This positive aspect of drug therapy highlights the importance of early diagnosis and prompt intervention to alleviate cardiac complications.
However, the therapeutic domain is not without challenges. Despite the benefits of ERT, its efficacy can vary significantly among patients, which may be influenced by factors such as the presence of anti-drug antibodies, timing of initiation and individual genetics. Additionally, some patients may experience infusion-related reactions and hypersensitivity, which may complicate treatment compliance. In addition, the high cost of ERT presents significant economic challenges to health systems and patients, raising concerns about equitable access to effective treatments.
The development of alternative pharmacological strategies has emerged in response to the limitations of ERT. Companion therapy, using small molecules to stabilize misfolded enzymes, has shown potential in early clinical trials. Pharmacological/ factorial approaches aimed at targeting pathways involved in lysosomal function and cellular homeostasis have the potential to provide additional treatments for controlling cardiac manifestations in patients with Fabry disease.
Additionally, ongoing research into gene therapy highlights the possibility of correcting the underlying genetic defect, providing the potential for a permanent cure. Advances in this area could transform the treatment approach for Fabry disease and its cardiac complications by addressing the root cause rather than simply controlling symptoms.
Conversely, the toxic effects of cumulative Gb3 are associated with myocardial ischemia and arrhythmias, contributing to the adverse cardiac events observed in patients with Fabry disease. Recognizing these negative effects is essential to understanding the full spectrum of disease and customizing treatment strategies accordingly. In addition, individuals with Fabry disease are at increased risk for cardiovascular events, including heart failure and sudden cardiac death, which complicate their clinical management and require an innovative approach that includes cardiology, genetics and pharmacology.
In summary, the cardiac pharmacology of Fabry disease illustrates the complex exchange between historical knowledge and contemporary advances in treatment. Although enzyme replacement therapy has significantly improved cardiovascular outcomes for many patients, persistent challenges remain, such as variability in treatment response, side effects and economic limitations. Research into alternative therapies and potential cures provides hope for a better future for those affected by this debilitating disease. Overall, the understanding of pharmacological interventions for Fabry disease not only highlights the urgency of early recognition and treatment but also highlights the need for continued research and innovation in the field of rare Fabry disease.
Citation: Carter J (2024). Cardiac Pharmacological Aspects Involved in Fabry Disease. Cardiovasc Pharm. 13:401.
Copyright: © 2024 Carter J. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.