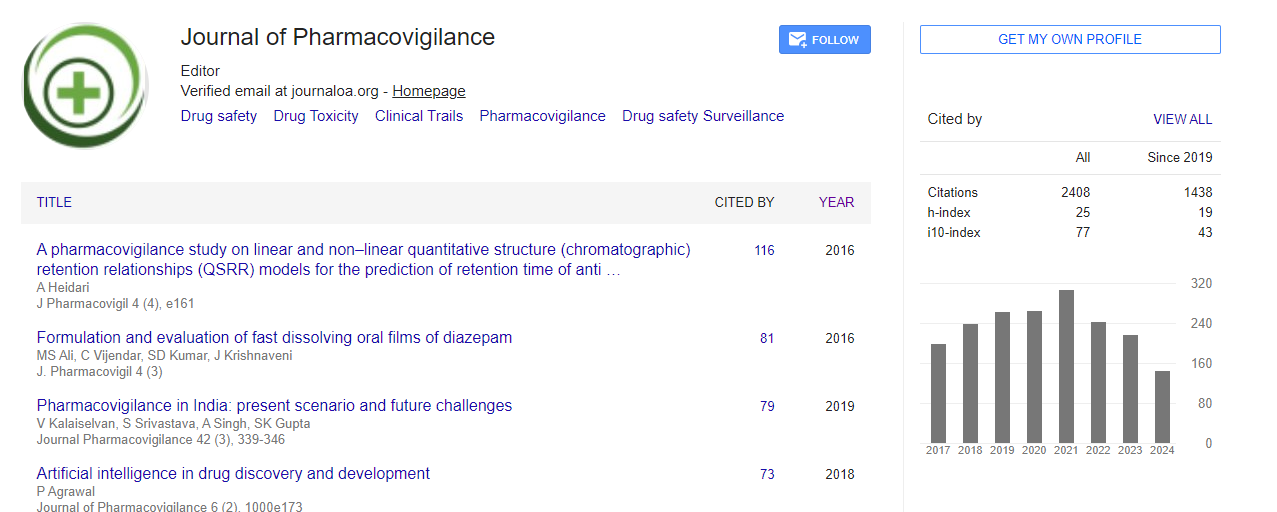

Indexed In
- Open J Gate
- JournalTOCs
- The Global Impact Factor (GIF)
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Mini Review - (2021) Volume 9, Issue 10
An Outline on Drug Designing, Targets and Computer-Aided Drug Designing
Stella Morris*Received: 29-Sep-2021 Published: 19-Oct-2021, DOI: 10.35248/2329-6887.21.9.340
noheading
Medication configuration, regularly alluded to as levelheaded medication plan or just judicious plan, is the innovative course of finding new prescriptions dependent on the information on a natural target. The medication is most usually a natural little particle that enacts or hinders the capacity of a biomolecule like a protein, which thus brings about a remedial advantage to the patient. In the most fundamental sense, drug configuration includes the plan of particles that are corresponding fit and charge to the biomolecular focus with which they collaborate and subsequently will tie to it. Medication plan much of the time however not really depends on PC displaying techniques. This kind of demonstrating is now and again alluded to as PC supported medication plan. At last, drug plan that depends on the information on the three-dimensional construction of the biomolecular target is known as construction based medication design. Notwithstanding little particles, biopharmaceuticals including peptides and particularly remedial antibodies are an undeniably significant class of medications and computational techniques for working on the proclivity, selectivity, and strength of these protein-based therapeutics have likewise been created.
The expression "drug configuration" is somewhat a misnomer. A more precise term is ligand plan (i.e., plan of an atom that will tie firmly to its target). Although plan methods for forecast of restricting partiality are sensibly fruitful, there are numerous different properties, like bioavailability, metabolic half-life, secondary effects, and so forth, that initially should be enhanced before a ligand can turn into a protected and effectual medication. These different attributes are regularly hard to foresee with objective plan methods. All things considered, because of high weakening rates, particularly during clinical periods of medication improvement, more consideration is being centered from the get-go in the medication configuration process around choosing applicant tranquilizes whose physicochemical properties are anticipated to bring about less difficulties during advancement and thus bound to prompt a supported, promoted drug. Furthermore, in vitro explores supplemented with calculation techniques are progressively utilized in early medication revelation to choose compounds with better ADME (ingestion, dispersion, digestion, and discharge) and toxicological profiles [1].
Drug Targets
A biomolecular target (most ordinarily a protein or a nucleic corrosive) is a key particle engaged with a specific metabolic or flagging pathway that is related with a particular sickness condition or pathology or to the infectivity or endurance of a microbial microorganism. Potential medication targets are not really infection causing however should by definition be illness modifying. At times, little particles will be intended to upgrade or hinder the objective capacity in the particular sickness altering pathway. Little atoms (for instance receptor agonists, enemies, reverse agonists, or modulators; compound activators or inhibitors; or particle channel openers or blockers) will be planned that are corresponding to the limiting site of target. Small particles (drugs) can be planned so as not to influence some other significant "askew" atoms (frequently alluded to as anti-targets) since drug communications with off-target atoms might prompt bothersome side effects. Due to similitudes in restricting destinations, firmly related targets recognized through arrangement homology have the most noteworthy possibility of cross reactivity and consequently most elevated aftereffect potential. Most ordinarily, drugs are natural little particles created through substance union, yet biopolymerbased medications (otherwise called biopharmaceuticals) delivered through organic cycles are turning out to be progressively more common. What's more, mRNA-based quality hushing advances might have restorative applications [2].
Rational Drug Discovery
Rather than conventional techniques for drug disclosure (known as forward pharmacology), which depend on experimentation testing of compound substances on refined cells or creatures, and coordinating with the evident impacts to medicines, levelheaded medication configuration (likewise called invert pharmacology) starts with a speculation that regulation of a particular natural objective might have helpful worth. All together for a biomolecule to be chosen as a medication target, two fundamental snippets of data are required. The first is proof that balance of the objective will be illness adjusting. This information might come from, for instance, sickness linkage concentrates on that show a relationship between changes in the organic objective and certain infection states. The second is that the objective is "druggable". This implies that it is equipped for restricting to a little particle and that its movement can be balanced by the little molecule. When an appropriate objective has been distinguished, the objective is typically cloned and delivered and purged. The refined protein is then used to set up a screening examine. Furthermore, the threedimensional design of the objective not really set in stone.
The quest for little atoms that tight spot to the objective is started by screening libraries of potential medication compounds. This might be finished by utilizing the screening measure (a "wet screen"). Furthermore, if the construction of the objective is accessible, a virtual screen might be performed of applicant drugs. In a perfect world the competitor drug mixtures ought to be "drug-like", that is they ought to have properties that are anticipated to prompt oral bioavailability, satisfactory substance and metabolic dependability, and negligible poisonous effects. Several techniques are accessible to assess druglikeness, for example, Lipinski's Rule of Five and a scope of scoring strategies, for example, lipophilic efficiency. Several techniques for foreseeing drug digestion have additionally been proposed in the logical literature. Because of the enormous number of medication properties that should be at the same time improved during the plan cycle, multi-objective enhancement procedures are now and again employed. Finally in view of the impediments in the current techniques for expectation of action, drug configuration is still especially dependent on serendipity and limited rationality [3].
Computer-Aided Drug Design
The most crucial objective in drug configuration is to foresee whether a given particle will tie to an objective and assuming this is the case how emphatically. Sub-atomic mechanics or sub-atomic elements is regularly used to gauge the strength of the intermolecular collaboration between the little particle and its natural objective. These strategies are likewise used to anticipate the adaptation of the little atom and to display conformational changes in the objective that might happen when the little particle ties to it. Semi-exact, abdominal muscle initio quantum science techniques, or thickness utilitarian hypothesis are regularly used to give upgraded boundaries to the sub-atomic mechanics computations and furthermore give a gauge of the electronic properties (electrostatic potential, polarizability, and so forth) of the medication up-and-comer that will impact restricting affinity. Atomic mechanics techniques may likewise be utilized to give semi-quantitative forecast of the limiting proclivity. Likewise, information based scoring capacity might be utilized to give restricting proclivity gauges. These strategies utilize direct relapse, AI, neural nets or other factual procedures to determine prescient restricting proclivity conditions by fitting test affinities to computationally inferred collaboration energies between the little particle and the target.
In a perfect world, the computational technique will actually want to foresee proclivity before a compound is orchestrated and subsequently in principle just one compound should be combined, saving gigantic time and cost. Actually present computational strategies are defective and give, best case scenario, just subjectively precise appraisals of liking. By and by it actually takes a few cycles of plan, combination, and testing before an ideal medication is found. Computational techniques have sped up disclosure by lessening the quantity of emphasess required and have regularly given novel designs [4].
REFERENCES
- Blackshields, Caroline A., Crean Abina M. Continuous Powder Feeding For Pharmaceutical Solid Dosage Form Manufacture: A Short Review. Pharm Deve Technol. 2018; 23 (6): 554â??560.
- Wang Yifan , Li Tianyi, Muzzio Fernando J., Glasser Benjamin J. Predicting Feeder Performance Based On Material Flow Properties. Powder Techn. 2017; 308: 135â??148.
- Cartwright James J , Robertson John , D'Haene Dorie , Burke Matthew D. , Hennenkamp Jeffrey R. Twin Screw Wet Granulation: Loss In Weight Feeding Of A Poorly Flowing Active Pharmaceutical Ingredient. Powder Techno. 2011. 238: 116â??121.
- Baxter Thomas, Prescott James. Developing Solid Oral Dosage Forms. Academic Press. 2009
Citation: Morris S (2021) An Outline on Drug Designing, Targets and Computer-Aided Drug Designing. J Pharamacovigil 9:340. DOI: 10.24105/2329-8790.2021.9.340
Copyright: © 2021 Morris S. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.