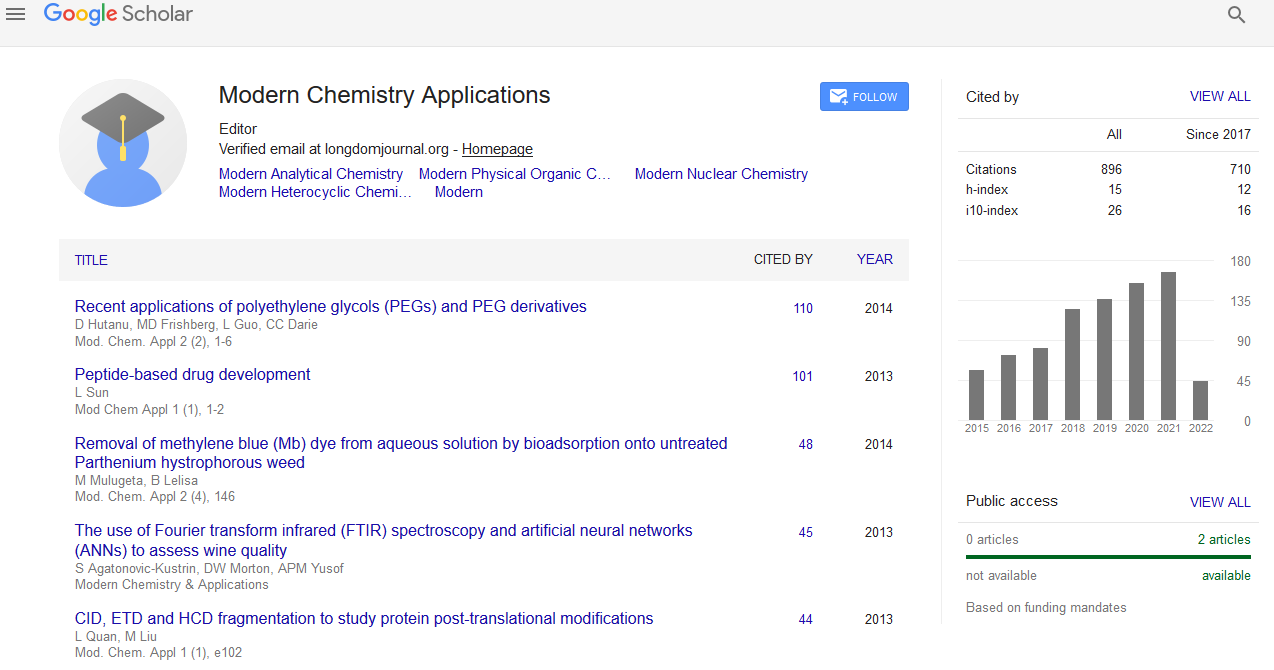

Indexed In
- Open J Gate
- JournalTOCs
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Scholarsteer
- Publons
- Geneva Foundation for Medical Education and Research
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2025) Volume 13, Issue 1
5-Nitrofuranyl Derivatives Shape Shift in Polar Aprotic Solvents Which May Give Rise to Induced Ring Currents in an Applied Magnetic Field
Ashley L. Dey*Received: 25-Aug-2023, Manuscript No. MCA-23-22724; Editor assigned: 28-Aug-2023, Pre QC No. MCA-23-22724 (PQ); Reviewed: 11-Sep-2023, QC No. MCA-23-22724; Revised: 09-Jan-2025, Manuscript No. MCA-23-22724 (R); Published: 16-Jan-2025, DOI: 10.35248/2329-6798.25.13.486
Abstract
Anisotropically induced ring currents are not confined to Hückel’s aromatics, and many non-aromatic compounds, in particular, cyclopropane exhibit ring currents in an applied magnetic field. To this end, aromatic protons of 5-nitrofurfural give rise to two signals in its 1H NMR spectrum in CDCl3 which as expected, are only ~ ± 0.1 ppm apart; however, in marked contrast to 5-nitrofurfural, the analogous aforementioned protons of (E)-5-nitrofuran-2-yl methylene hydrazine are nearly ± 1.0 ppm apart in its 1H NMR spectrum in DMSO-d6. This anomaly may, though it need not of necessity, suggest that the hydrazone moiety in (E)-5-nitrofuran-2-yl methylene hydrazine force the furan ring into a puckered non-aromatic ring so as to pave the way for an induced ring current in an applied magnetic field. 5-nitro-N'-5-nitrofuran-2-carbonyl furan-2-carbohydrazide also appears to exhibit a similar perturbation; however, to a lesser extent.
Keywords
(E)-5-nitrofuran-2-yl methylene hydrazine; 5-nitro-N'-5-nitrofuran-2-carbonyl furan-2-carbohydrazide; Extended π-delocalisation; 1H NMR of nitrofuranyl derivatives; Heterocycles; Shapeshifting; Molecular recognition; Induced ring currents
Introduction
A regular 5-membered planar carbocyclic ring has an internal valence angle of 108°, though almost free of Baeyer strain, such a conformation creates a sizeable Pitzer strain (~42 kJ mol-1 in virtue of 10 pairs of eclipsing H-H) such that a regular cyclopentane becomes puckered, thereby reducing the sum of Pitzer and Baeyer strain to ~60% of that for a planar conformation. Thus, the most stable conformations of a regular cyclopentane are envelope and half-chair (Figure 1), with the energy barrier between the two conformations being negligible i.e. 2.1 kJ mol-1, a regular cyclopentane will not even pass the high energy planar conformation in a supposed ‘conformational flux.’
Kilpatrick et al. recognised that the envisaged ‘conformational flux’ in a cyclopentane arises from oscillation of its carbons perpendicular to the plane of the ring, and thereupon imitating a rotating “bulge” (out-of-plane atom) around the ring, which has been referred to as ‘pseudorotation’, even though, in point of fact, there is no movement in that direction [1]. To this end, for an asymmetric cyclopentane the aforementioned ‘pseudorotation’ gives rise to a total of 20 unique energy minima (10 envelopes and 10 half-chairs), and an infinite number of quanta in between the aforementioned minima thereof.

Figure 1: Major conformations of 5 and 6-memebered rings: a) The most stable conformations of a regular cyclopentane; b) The global minimum (left), and maximum (right) conformations of cyclohexane; c) The most stable conformation of tetrahydrothiophene.
Consequently, unlike cyclohexane for which its lowest energy minimum i.e., chair conformation is located in a deep energy valley, and with a conformational flux interconversion energy barrier of 44.8-48.1 kJ mol-1 (Figure 1) between the chair i.e. global minimum and the half-chair i.e. global maximum conformations being nearly 20 times that of its cyclopentane counterpart, the preferred conformation for a substituted cyclopentane is one that minimises the sum of Pitzer and Baeyer strain in a manner that both interactions between the substituents on the ring and the substituents themselves with the ring are kept at the absolute minimum [2]. This phenomenon is also true for a regular cyclopentene, and also cyclopentadiene even so with considerably less strain than both a regular cyclopentane and cyclopentene, alongside most of their corresponding heterocyclic counterparts; in point of fact, tetrahydrothiophene which is isoelectronic with Tetrahydrofuran (THF), exists exclusively in its half-chair conformation with the thioether moiety in the least puckered middle region.
Materials and Methods
Materials and instruments
All reagents and solvents in the experiments were of reagent grade quality. They were obtained from commercial sources and used without further purification; unless otherwise stated. Diethyl ether herein is referred to as ether. Water used was from a Millipede Milli-Q EDM Water Purification A10 unit or was HPLC grade. High performance Liquid Chromatography-Mass Spectrometry (LC-MS) analyses were performed using an Agilent UPLC SQD instrument equipped with a Chromolith®, C18, 2.1 × 50 mm column. The eluent was 0.1% formic acid (v/v) in water (solvent A)/ acetonitrile (solvent B) over a 5-95% acetonitrile gradient; monitoring at 214 nm over 10 min. Electrospray ionisation mass spectrometry was performed using an Agilent single quadrupole unit equipped with CTC-PAL. High-resolution electrospray mass spectra were obtained on a Waters LCT Premier XE spectrometer [3]. Melting points were determined using a Gallenkamp MFG 595 010 melting point apparatus, and a Reichert Austria hot stage microscope. 1H and 13C NMR spectra were recorded on a Bruker AVIII HD 400 unit equipped with prodigy N2 broadband CryoProbe operating at 400 MHz and 101 MHz, respectively, using CDCl3, D2O, methanol-d4, acetonitrile-d3 and dimethyl-d6 sulfoxide (DMSOd6) as solvents. Chemical shifts (δ) are given in parts per million (ppm) relative to the internal Tetramethylsilane (TMS). Coupling constants are given in Hz. The abbreviations s, d, and m correspond to singlet, doublet, and multiplet, respectively. Reaction products were identified by their NMR, and/or otherwise by LC-MS and HRMS analyses [4].
(E)-(5-Nitrofuran-2-yl)methylene hydrazine
5-Nitrofurfural (250 mg, 1.8 mmol) and anhydrous Na2SO4 (50 mg) were placed in a flame-dried reaction vessel, sealed and degassed under argon. Thereto was added anhydrous THF (1.0 ml) and stirred for 2.0 min, followed by addition of hydrazine monohydrate (0.10 ml, 2.0 mmol), and the thus obtained red reaction mixture was stirred under argon at ambient temperature for 24 h. Upon completion TLC controlled (5.0% methanol in CH2Cl2; Rf 0.20) the dark red cloudy reaction mixture was diluted with deionised water (5.0 ml), and extracted with EtOAc (5.0 × 25 ml). The extracts were combined and dried over anhydrous Na2SO4, filtered and the solvent was removed in vacuo to afford crude hydrazone 1 (250 mg; 90%) as a red solid, which was further purified by flash column chromatography (EtOAc/cyclohexane, 1:5) to afford the desired product 1 (100 mg; 36%) as needle-like orange crystals; Rf 0.25 (80% EtOAc in cyclohexane); mp 153â??–154â?? (from acetonitrile) (lit.i 164â?? dec.); δH (400 MHz, acetonitrile-d3) 7.58 (1 H, s, CCHN unsaturated), 7.43 (1 H, d, J 3.9, CH-Ar),6.66 (1 H, d, J 3.9, CH-Ar); δC (101 MHz, DMSO-d6) 108.8 (CH-Ar), 116.1 (CH-Ar), 123.7 (CCHN unsaturated); LC-MS (retention time: 3.87 min) m/z (ESI) 156 (M+ + H, 100%); HRMS (ESI) calcd. for C5H5N3O3 (M+H)+ m/z 156.0404, found 156.0403.
5-Nitro-N'-(5-nitrofuran-2-carbonyl) furan-2- carbohydrazide
5-Nitro-2-furonyl chloride (0.18 g, 1.0 mmol) and anhydrous Na2SO4 (50 mg) were placed in a flame-dried reaction vessel, sealed and degassed under argon. Thereto was added anhydrous THF (10 ml) and stirred for 2.0 min, followed by addition of hydrazine monohydrate (49 μl, 1.0 mmol), and the thus obtained red reaction mixture was stirred under argon at 20â?? overnight. Upon completion-TLC controlled (60% EtOAc in cyclohexane; Rf 0.25) the dark red cloudy reaction mixture which was acidic to Congo red, was diluted with deionised water (5.0 ml), and washed with EtOAc (5.0 × 25 ml) [5]. Thereto was added saturated NaHCO3aq to adjust the pH to ~ 7.0 (caution!effervescent), and the aqueous was extracted with EtOAc (6.0 × 25 ml). The extracts were combined and dried over anhydrous Na2SO4, filtered and the solvent was removed in vacuo to afford crude carbohydrazide 2 (250 mg; 81%) as a dark orange solid, which was further purified by flash column chromatography (EtOAc/cyclohexane, 2:1) to afford the desired product 2 (200 mg; 65%) as a coral pink solid; Rf 0.25 (80% EtOAc in cyclohexane); mp 180–185â?? dec. (from methanol); δH (400 MHz, DMSO-d6) 7.74 (2 H, d, J 3.9, 2 × CHAr), 7.34 (2 H, d, J 3.9, 2 × CH-Ar); δC (101 MHz, DMSO-d6) 113.8–114.9 (2 × CH-Ar), 149.9–151.3 (2 × C-Ar), 154.0 (C=O carbohydrazide); LC-MS (retention time: 5.08 min) m/z (ESI) 311 (M+ + H, 100%), 172 (50), 141 (16).
Results and Discussion
As part of a medicinal chemistry endeavour aimed at the development of next-generation antibacterial drug conjugates (unpublished), hydrazone 1 was synthesised from 5-nitrofurfural and hydrazine monohydrate in anhydrous acetonitrile in 62% yield (cf. Experimental); however this heterocycle either reacts with methanol, water and most protic solvents, or is virtually insoluble in most common organic solvents such as ether, CH2Cl2, chloroform, toluene etc., such that restricts the solvent of choice for its NMR measurements to either acetonitrile-d3 in which 1 is sparingly soluble or DMSO-d6 which itself is quite hygroscopic in nature. Nevertheless, a relatively clean 1H NMR spectrum of 1 was obtained in acetonitrile-d3 (Figure 2) for which in marked contrast to that of 5-nitrofurfural in CDCl3, the signal from one of the aromatic protons in 1 has been upshifted by almost 0.75 ppm relative to that from the other aromatic proton in the same spectrum; this effect is even more pronounced for 1H NMR spectrum of 1 in DMSO-d6 which is not applicable to that of 5-nitrofurfural (Figure 3) [6 ].

Figure 2: a) 1H NMR spectrum of 1 in acetonitrile-d3; b) 1H NMR spectrum of 5-nitrofurfural in CDCl3.

Figure 3: 1H NMR spectrum of 1 (top) stacked over that of 5-nitrofurfural (bottom) in DMSO-d6; (the pertinent aromatic region is magnified in the blue oval).
Reference has already been made to the ground state conformations of 5-memebered heterocycles, in particular, tetrahydrothiophene which is isoelectronic with THF (vide supra); to this end, it is conceivable that the addition of hydrazine to 5-nitrofurfural renders a ‘pseudoumpolung’ whereby the ensuing plausible conjugative effect pushes the furan ring into a similar non-planar puckered heterocyclopentene conformation, facilitating an extended π-delocalisation (Figure 4); in essence, setting up a weak induced ‘ring current’ in complexation with DMSO-d6 in an applied magnetic field, such that one of the protons on the furan ring falls inside the ring, and thence, upshifted at ca. δ 6.7 in 1H NMR spectrum of 1 in DMOS-d6; on the contrary, the other proton sits outside the anisotropically induced ring current and downshifted at ca. δ 7.6 relative to its counterpart, pushing the signals from these two protons in the aforementioned 1H NMR spectrum of 1 almost ± 1.0 ppm apart, which are otherwise not chemically too different in say, 5-nitrofurfural [7].

Figure 4: It can be hypothesised that the extended electron shuffle assisted by DMSO-d6 shapeshifts the furan ring into a puckered ring so as to set up an induced ring current (purple) in the applied magnetic field, resulting in ~± 1.0 ppm shift between the signals from the pertinent protons (light blue) in 1H NMR spectrum of 1.
It cannot be too firmly emphasised, however, that similar but different to 1, it appears as though in solution, especially in a DMSO medium ~5.0–10% of homodimer 3 tautomerises to form heterodimer 4 (Figure 5) by virtue of complexation with the polar solvent i.e. DMSO [8 ].

Figure 5: Further tautomerisation of homodimer 3 yields heterodimer 4.
Support for the above phenomenon can be provided by the observation that similar to 1, the enol part of heterodimer 4 can now push the furan ring into a puckered ring so as to set up a weak induced ring current in an applied magnetic field by virtue of complexation with DMSO-d6 (Figure 6). It will thus become apparent as to why the minor signals in the 1H NMR spectrum of this nitrofuran at ca. δ 7.6 and 6.9 are exactly 0.7 ppm apart, nearly double of that observed for the major signals at ca. δ7.7 and 7.3 (Figure 7). This large shift between the minor signals can be explained by the aforementioned induced ring current.

Figure 6: It can be hypothesised that 4 sets up a weak induced ring current in an applied magnetic field by virtue of complexation with DMSO-d6.
Unfortunately, the above dimer is virtually insoluble in relatively less polar solvents such as chloroform, dichloromethane, diethyl ether etc., so as to compare the solvent effects on the position of 1H NMR signals from the protons of furan rings in the dimer, and validate the supposedly induced ring current; nevertheless, this effect has clearly been echoed by 1, and it may, though it need not of necessity, be safe to conclude that in the light of its 1H NMR spectrum in DMSO-d6, this dimer should also follow suit [9].

Figure 7: 1H NMR spectrum of homodimer 3, illustrating a large shift i.e. 0.7 ppm between minor signals at δ 7.58-7.57 and 6.88-6.87, presumably due to complexation with DMSOd6 to set up a weak induced ring current in the applied magnetic field.
Conclusion
The signals from the aromatic protons in 1H NMR spectra of 5- nitrofufural in polar aprotic deuterated solvents, in particular, DMSO-d6 are in stark contrast to that of 1 and 2. This anomaly, on looking more closely, can be explained by means of an anisotropically induced ring current facilitated by complexation with the polar solvent molecules such that the thus obtained furan ring is no longer planar and has been shapeshifted into a puckered ring thereby lowering the energy of the entire system, and that this should be so may, in part, be a corollary of the well-established conjugative effect. Whether this anomaly stems from an induced ring current or is merely a corollary of proximity-induced displacements, further magnetic susceptibility measurements and/or DFT calculations, on the basis of the work herein, can provide support for or against this puckered nitrofuran hypothesis.
Conflict of Interest
The author declares no conflicts of any kind.Acknowledgment
This work was supported by the biotechnology and biological sciences research council (Grant No. BB/P504373/1) and GlaxoSmithKline. The author would like to extend sincere gratitude to the royal society of chemistry for their generous donations facilitated by Kim Sutton. Special thanks are also due to Professor M. G. Moloney for helpful discussions.
References
- Aston JG, Schumann SC, Fink HL, Doty PM. The structure of alicyclic compounds. J Am Chem Soc. 1941;63(7):2029-2030.
- Pitzer KS, Donath WE. Conformations and strain energy of cyclopentane and its derivatives. J Am Chem Soc. 1959;81(13):3213-3218.
- Kilpatrick JE, Pitzer KS, Spitzer R. The thermodynamics and molecular structure of cyclopentane1. J Am Chem Soc. 1947;69:2483-2488.
- Kellie GM, Riddell FG. Nonâ?Chair Conformations of Sixâ?Membered Rings. Top Stereochem. 1974:225-269.
- JD Cox, G Pilcher. Thermochemistry of Organic and Organometallic Compounds. Academic, New York. 1970.
- Wiberg KB, Bonneville G, Dempsey R. Strain Energies of Small Ring Alkenes. Israel J Chem. 1983;23(1):85-92.
- Wiberg KB, Walker FH, Pratt WE, Michl J. [2.1. 1]. Propellane. Reaction of 1, 4-diiodobicyclo (2.1. 1) hexane with tert-butyllithium and with potassium atoms. J Am Chem Soc. 1983;105(11):3638-3641.
- Fuchs B. Conformations of Fiveâ?Membered Rings. Top Stereochem. 1979:1-94.
- FG Riddell. The Conformational Analysis of Heterocyclic Compounds. Academic, New York. 1980.
Citation: Dey AL (2025) 5-Nitrofuranyl Derivatives Shape Shift in Polar Aprotic Solvents Which may Give Rise to Induced Ring Currents in an Applied Magnetic Field. Modern Chem Appl. 13:486.
Copyright: © 2025 Dey AL. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.